
Sugestão de condutas para o tratamento dos neurofibromas plexiformes
Nilton Alves de Rezende (1)
Luiz Oswaldo Carneiro Rodrigues (1)
Juliana Ferreira de Souza (1)
Bruno Cezar Lage Cota (1)
Luíza de Oliveira Rodrigues (1, 2)
Instituições
- Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais
- CASU – Coordenadora de Avaliação de Novas Tecnologias em Saúde
Introdução
As neurofibromatoses atingem cerca de 80 mil brasileiros e sabemos que os neurofibromas plexiformes (NP) acometem cerca de 50% das pessoas com Neurofibromatose do tipo 1 (NF1) (Jiang e col., 2015), os quais surgem na vida intrauterina em qualquer parte do corpo e são de evolução imprevisível.
Os NP são a principal causa de morte (transformação maligna) e de complicações entre as pessoas com NF1 (dor, deformidades estéticas e perdas funcionais) (Riccardi, 2010).
Sabe-se que os NP são de crescimento lento, mas resistentes à quimioterapia e radioterapia tradicionais (Robertson e col., 2012), mas nos últimos 5 anos alguns estudos indicaram que metade dos NP reduziram seu volume (cerca de 30%) com uma droga chamada selumetinibe (ver aqui mais informações).
No entanto, para a maioria dos NP, sejam eles do tipo epineurais (difusos), assim como para os perineurais (nodulares) (Riccardi, 2007), o tratamento, quando necessário (por sintomas – ver abaixo) continua sendo cirúrgico.
Veja revisão recente do cirurgião professor José Renan da Cunha Melo na edição comemorativa dos 20 anos do nosso Centro de Referência em Neurofibromatoses (CRNF) do Hospital das Clinicas da Universidade Federal de Minas Gerais (clique aqui).
A conduta médica deve ser definida em cada pessoa em função da localização do NP, dos seus impactos na qualidade de vida da pessoa, da sua taxa de crescimento, dos sintomas que produz (dor, deformidade, disfunção) e do risco de transformação maligna.
Diante de cada uma dessas situações, deve ser pesada a viabilidade da cirurgia e o seu respectivo risco cirúrgico.
O risco cirúrgico dos NP é sempre considerável, pois, além das dificuldades técnicas de cada caso (localização, estruturas vitais envolvidas), os NP costumam sangrar muito durante o procedimento cirúrgico e para isto o banco de sangue deve estar de sobreaviso especial.
Além disso, a dor neuropática pode ser uma sequela da cirurgia (ver aqui mais informações sobre a dor neuropática).
A Tabela 1 apresenta uma sugestão de conduta para os cinco níveis de estado clínico dos NP: (1) estável, (2) crescendo sem sintomas, (3) crescendo com sintomas (dor, disfunção neurológica, impacto estético), (4) crescendo com sintomas e risco de morte (sinais sugestivos de grande atividade celular, por exemplo, captação aumentada de glicose no PET CT) e (5) transformação maligna evidente.
Tabela 1 – Sugestão de condutas nos neurofibromas plexiformes nas pessoas com Neurofibromatose do Tipo 1. (Fotos de 1 a 6 no Anexo I ilustram casos dos níveis C e D)
| Estado clínico dos neurofibromas | |||||
| Risco da cirurgia |
1 Estável |
2
Crescendo |
3
Crescendo com sintomas |
4
Crescendo com sintomas e risco de morte |
5 Transformação maligna |
| A Baixo | Cirurgia | Cirurgia | Cirurgia | Cirurgia | Cirurgia |
| B Médio | Observar | Observar | Cirurgia | Cirurgia | Cirurgia |
| C Alto | Observar | Observar | Cirurgia? | Cirurgia | Cirurgia |
| D Inviável | Observar | Paliativo | Paliativo | Radioterapia? | Radioterapia |
Do ponto de vista da cirurgia, podemos considerar quatro níveis de dificuldades técnicas: (A) baixo risco (tumores superficiais, relativamente bem delimitados, de fácil acesso), (B) médio risco (tumores internos, porém ressecáveis, sem envolver estruturas vitais), (C) alto risco (tumores profundos, envolvendo estruturas internas, especialmente torácicas, plexos nervosos e sistema vascular), e (D) inviável (tumores inoperáveis, geralmente de grande volume, difusos e envolvendo estruturas vitais).
Os PN sintomáticos (dor, deformidade e disfunção) e inoperáveis (na Tabela 1, as situações D3 e D4) constituem o maior problema para as pessoas com NF1, motivo pelo qual têm sido buscados tratamentos medicamentosos capazes de reduzir a dor, o volume e os seus impactos sobre a saúde.
Como crescem os neurofibromas plexiformes?
Para começar, precisamos lembrar a definição de neurofibromas plexiformes: são tumores benignos, geralmente congênitos (já estão presentes na criança dentro do útero), formados em vários feixes de nervos e que às vezes se estendem a outros órgãos ou tecidos ao redor.
Geralmente, os NP apresentam mais de 3 cm de diâmetro, enquanto os neurofibromas cutâneos raramente atingem 3 cm de tamanho.
Eles podem surgir e se tornarem visíveis na cabeça, pescoço, tronco ou extremidades (braços e pernas), ou ainda se formarem profundamente nessas mesmas regiões sem serem visíveis.
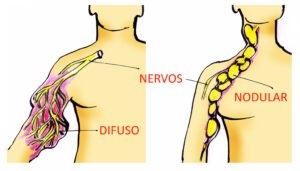
Em 100 pessoas com NF1, cerca de 50 delas apresentam neurofibromas plexiformes, sendo metade (25) do tipo difuso (que são macios, pouco definidos e de formas irregulares) e metade (25) do tipo nodular (firmes, bem definidos e de forma ovalada).
Uma mesma pessoa pode apresentar ambos os tipos de neurofibromas.
Para dizer se um medicamento é eficiente ou não para tratar os neurofibromas, temos que saber como os neurofibromas se comportam sem qualquer tratamento.
Isto se chama história natural dos neurofibromas: como aparecem, como crescem, quais os sintomas e problemas que produzem.
Consideramos que um neurofibroma está em crescimento quando ele aumenta em volume mais do que a taxa normal de crescimento corporal da criança.
Então, nossas perguntas são:
- Quando aparecem os neurofibromas plexiformes?
- Os plexiformes sempre aumentam de tamanho, permanecem estáveis ou diminuem com o passar do tempo?
- Qual é a taxa de crescimento anual dos neurofibromas? Neurofibromas plexiformes difusos se comportam da mesma forma que neurofibromas nodulares?
- A idade, sexo e raça da pessoa com NF1 influencia o crescimento dos neurofibromas plexiformes?
Um estudo muito interessante sobre algumas destas questões foi publicado em março de 2020 pela Dra. Srivandana Akshintala em colaboração com o grupo da pesquisadora norte-americana Brigitte Widemann, o mesmo grupo que testou o selumetinibe e cujos resultados serviram de base para a aprovação do medicamento pela FDA nos Estados Unidos.
Uma tradução adaptada do título deste estudo pode ser: “Avaliação do crescimento dos neurofibromas plexiformes nodulares e difusos ao longo dos anos, em crianças e adultos jovens com NF1.” (ver o trabalho completo aqui, em inglês: https://pubmed.ncbi.nlm.nih.gov/32152628/ )
As ideias para este estudo surgiram há cerca de 15 anos e, entre 2008 e 2015, 138 pessoas com NF1 e neurofibromas plexiformes inscreveram-se como voluntárias e foram acompanhadas por cerca de 6 anos, com avaliações clínicas periódicas e medida do volume dos tumores por ressonância magnética. Ao final, os dados de 122 pessoas puderam ser analisados.
Os resultados do estudo da Dra. Akshintala permitiram responder a algumas das perguntas que mencionamos acima:
- Quando aparecem?
Os neurofibromas difusos são diagnosticados precocemente na infância (primeira ressonância em geral é realizada em torno dos 8 anos) e os neurofibromas nodulares são diagnosticados geralmente mais tarde (primeira ressonância em torno dos 15 anos).
- Como se comportam?
Há neurofibromas que apresentam períodos de crescimento, outros apresentam fases de estabilidade e cerca de 14 em cada 100 deles podem regredir em tamanho (sem qualquer tratamento).
Neste estudo da Dra. Akshintala e colaboradores, a maioria dos tumores cresceu durante o período de observação:
85 em cada 100 plexiformes difusos, e 76 em cada 100 plexiformes nodulares aumentaram de volume
- Qual a taxa de crescimento?
A taxa de crescimento varia muito entre diferentes pessoas com NF1 e entre dois plexiformes na mesma pessoa, mas cada tumor apresenta uma certa constância de crescimento na mesma pessoa.
Além disso, a taxa de crescimento foi muito diferente entre nodulares e difusos: os plexiformes difusos cresceram 19% ao ano e os plexiformes nodulares cresceram 44% ao ano.
- Idade, o sexo e a raça afetam os plexiformes?
A idade, sim, pois as maiores taxas de crescimento dos plexiformes difusos ocorrem na infância antes dos 5 anos de idade.
Já os neurofibromas nodulares não dependem muito da idade, mas parecem crescer mais a partir da adolescência.
Não foram percebidos efeitos importantes do sexo nem houve diferença entre brancos e não-brancos na taxa de crescimento dos plexiformes.
Conclusão
Por enquanto, nossa compreensão sobre os NP é a seguinte:
- Neurofibromas plexiformes difusos parecem ter uma biologia diferente dos nodulares.
- A maioria dos difusos cresce na infância e os nodulares crescem mais tarde.
- Um em cada dez neurofibromas plexiformes pode regredir espontaneamente sem tratamento.
A equipe do CRNF reuniu recomendações sobre o cuidado com os neurofibromas plexiformes sintomáticos e inoperáveis, incluindo o uso ou não do medicamento selumetinibe CLIQUE AQUI PARA VER O PROTOCOLO DE CUIDADOS DO CRNF

Como saber se o tumor é benigno ou maligno?
Nas pessoas com neurofibromatose do tipo 1 (NF1), frequentemente precisamos esclarecer se um tumor é um neurofibroma ou se este neurofibroma sofreu uma transformação maligna, o que ocorre em cerca de 5 a 10% dos neurofibromas plexiformes e nodulares.
Essa dúvida ocorre quando uma pessoa com NF1 apresenta um tumor, um neurofibroma plexiforme ou um neurofibroma nodular – já existente, que cresceu rapidamente ou mudou de consistência (ficou mais duro e firme) ou começou a doer (dor forte e persistente) ou atrapalhou a função neurológica ou mais de uma destas mudanças ao mesmo tempo.
Nessa situação acima, temos que agir rapidamente para esclarecer se continua sendo um neurofibroma benigno ou se uma parte dele sofreu transformação para Tumor Maligno da Bainha do Nervo Periférico (TMBNP).
Os TMBNP são tumores agressivos que precisam ser retirados cirurgicamente o mais breve possível, pois não respondem bem à quimioterapia e nem à radioterapia.
Diante dessa dúvida, quais são nossas condutas?
Resumimos abaixo algumas das condutas adotadas em nosso CRNF para tentarmos saber se um tumor é benigno ou maligno.
- Podemos realizar uma Biópsia
A retirada de uma parte do tumor (por agulha ou por cirurgia) e sua análise no laboratório seria o padrão mais confiável para definirmos se houve a transformação maligna, mas a biópsia apresenta alguns problemas nas pessoas com NF1.
A biópsia pode ser útil em outros tipos de câncer, quando todo o tumor é canceroso. Assim, qualquer fragmento colhido pela agulha poderá mostrar o tumor maligno.
Na NF1 não acontece assim. Nos neurofibromas, a transformação maligna geralmente ocorre em parte do tumor. Então, a biópsia pode colher um fragmento da parte maligna e confirmar o diagnóstico e ajudar a salvar a vida da pessoa.
No entanto, a biópsia por agulha pode pegar apenas um fragmento da parte que continua sendo neurofibroma e não revelar o grande perigo que está ao lado.
Portanto, nas pessoas com NF1, não confiamos na biópsia com agulha quando o resultado é negativo (ver mais informações aqui).
Além disso, para o acompanhamento de neurofibromas ao longo dos anos, biópsias repetidas podem não ser o método mais confortável para as pessoas. Então, precisamos contar com outros métodos que nos ajudem a esclarecer se um neurofibroma sofreu transformação maligna.
- Podemos recorrer aos métodos de diagnóstico por imagens
Em 2022, cientistas chineses (Liu e colaboradores) publicaram uma revisão dos métodos diagnósticos da transformação maligna de neurofibromas, incluindo aqueles disponíveis e outros em estudo, como a Inteligência Artificial (ver aqui o artigo completo, em inglês).
Apesar do número relativamente pequeno de estudos científicos, por causa da NF1 ser uma doença rara, há vantagens e desvantagens de cada método, que serão resumidas a seguir.
- Ultrassom
O ultrassom é um método não invasivo e econômico.
Quando um neurofibroma se torna maligno o ultrassom mostra tumores lobulados, com aspecto heterogêneo, com sinais de ampla circulação sanguínea.
No ultrassom de alta resolução, podem ser encontrados sete sinais sugestivos de transformação maligna:
- Tumores maiores do que 5 cm
- Crescimento rápido em semanas ou meses
- Margens irregulares com edema
- Aspecto heterogêneo
- Aspecto infiltrativo
- Vascularização estenótica, oclusiva, trifurcada e arcaica (enquanto nos benignos é hierárquica)
- Contraste aumentado na periferia e inalterado no centro do tumor
No entanto, pelo fato do ultrassom ser um método que exige a presença do médico para ser realizado, seu acesso é mais difícil do que a ressonância magnética ou a tomografia, que podem ser realizados por pessoas com formação técnica.
- Ressonância Nuclear Magnética (RNM)
A RNM é o método atualmente preferido para identificar tumores malignos, porque apresenta melhor resolução para tecidos moles, como são os neurofibromas.
Na RNM, nas imagens em T1, os neurofibromas benignos e o TMBNP são difíceis de distinguir, mas alguns achados ajudam a definir a possível malignidade:
- Sugestivos de TMBNP:
- Tamanho dos tumores maiores do que 5 cm
- Imagens em T1 com gadolínio com aumento periférico da intensidade no TMBNP (e central nos neurofibromas)
- Presença de hemorragia, necrose e aspecto cístico
- Tumores com margens borradas (“em penugem”) e com edema ao redor
- Sugestivos de neurofibromas benignos:
- Massas bem definidas com alta intensidade em T2
- Presença de área central menos intensa – o “sinal do alvo”
Com estes critérios, alguns estudos sugerem cerca de 65% de sensibilidade e 90% de especificidade para detecção do TMBNP.
A RNM funcional pode aumentar a sensibilidade para 88% e a especificidade para 94% se considerar os índices de difusão.
A RNM em 3 dimensões pode auxiliar na detecção precoce de aumentos volumétricos dos tumores e a arga tumoral aumentada na ressonância de corpo inteiro também pode sugerir a possibilidade de transformação maligna.
Limitações da RNM são:
- É um método dependente da subjetividade do examinador
- Há poucos estudos com os neurofibromas “atípicos” (tumores com padrão intermediário entre benigno e maligno)
- Tomografia computadorizada (TC)
Não dispomos ainda de critérios seguros para distinguir neurofibromas de TMBNP apenas com a TC. Alguns estudos estão associando a TC com aprendizado de máquina (inteligência artificial) e podem no futuro trazer uma contribuição a esta questão.
- Positron Emission Tomography Imaging (PET)
O método mais popular combina o PET com a CT (PET/CT), usando o contraste radioativo 18F-FDG.
Os achados mais importantes dos TMBNP no PET/CT são:
- Aumento da captação do contraste (SUV maior que 3,15)
- Heterogeneidade da captação
Limitações do 18FDG-PET/CT são:
- Sobreposição de SUV entre neurofibromas atípicos e TMBNP
- Sensibilidade 100% lesões assintomáticas – especificidade para negativo 100% – especificidade para positivo 46%
- Custo elevado
Alguns estudos estão sendo realizados com novos marcadores, como 68Ga-PSMA PET/CT e 11-Metionina, mas ainda não temos um consenso sobre seu uso nas pessoas com NF1.
- Combinação de PET com RNM (PET/RNM)
Ainda com poucos estudos em NF1 e sem disponibilidade em nosso meio.
- Radiogenômica
Trata-se de novos métodos em estudo, que procuram associar os resultados das imagens com análise genética. Alguns dos autores do estudo (Liu e outros) dividiram o gene NF1 em 5 domínios e associaram com dados clínicos e imagens de RNM e encontraram forte correlação com a transformação maligna.
Conclusão
Ainda não temos um consenso internacional, mas o indicador mais forte de transformação maligna é o aumento significativo do tamanho de um neurofibroma.
Nosso desafio futuro será encontrar correlações entre imagens e os 6 tipos histológicos de tumores mais comuns na NF1:
- Neurofibroma
- Neurofibroma com atipias celulares
- Neurofibroma celular
- ANNUBP – neurofibromas atípicos de potencial biológico incerto
- TMBNP de baixo grau
- TMBNP de alto grau
Por enquanto, precisamos combinar métodos e avançar nos estudos com inteligência artificial.
Belo Horizonte, 7 de janeiro de 2025