“Meu filho de 9 anos apresenta NF1 e escoliose. O que devo fazer?” JBS, de Natal, RN.
Cara J., obrigado pela sua pergunta, pois a escoliose é uma das alterações no desenvolvimento ósseo que ocorrem nas pessoas com NF1 com maior frequência (entre 10 e 60%) do que na população em geral (1 a 3%).
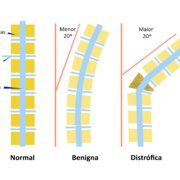
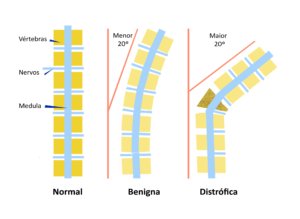
Escoliose significa qualquer curvatura da coluna maior do que 10 graus numa escala própria chamada de Escala de Cobb.
Geralmente, nas crianças com NF1, a escoliose aparece antes dos 14 anos de idade, mas raramente depois. Além disso, costuma se estabilizar ao longo da adolescência.
Quando a escoliose é simples, ou seja, apenas uma curvatura suave na coluna e o ângulo for menor do que 20 graus e não encontramos outras alterações ósseas, a evolução geralmente é benigna e bastam exercícios regulares, cuidados com a Vitamina D e observação clínica.
Porém, quando a escoliose é mais acentuada, ou seja, o ângulo da curvatura for maior do que 20 graus e, principalmente, se houver outras alterações ósseas, chamamos de escoliose distrófica, que apresenta maior chance de progressão e necessidade de tratamento cirúrgico.
Sintomas
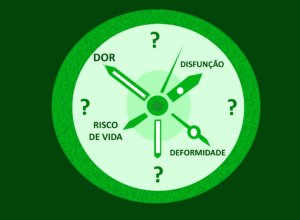
Os sintomas mais comuns na escoliose são dor em cerca de 20% das pessoas com NF1. Geralmente, a dor ocorre na coluna torácica (10%) e lombar (9%). Pode haver perda da função sensorial (5%) e motora (10%, mais comum no sexo masculino).
Alterações ósseas associadas com a NF1
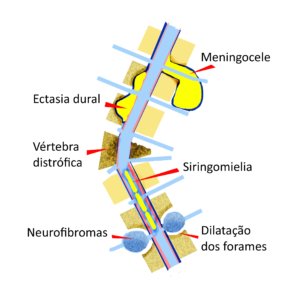
Nas pessoas com NF1, algumas alterações ósseas (ver figura) associadas à escoliose indicam a necessidade de maior cuidado médico :
- Deformidades das vértebras (cerca de 5%)
Podem ocorrer distrofias, ou seja, defeitos de formação, quando uma ou mais vértebras apresentam problemas estruturais como desabamento, fraturas, baixa densidade óssea, deformidades e falhas.
- Ectasia dural (45%)
Ectasia quer dizer “fora do lugar” e ocorre quando as membranas que envolvem a medula (chamadas de dura máter e aracnoide) apresentam dilatações e invadem o corpo das vértebras. Se aumentarem ao longo do tempo, precisam ser tratadas para evitar a progressão da escoliose.
- Erosões (20%)
Geralmente associadas com a ectasia dural, são defeitos de formação na parte central das vértebras (chamados de scalloping na ressonância magnética). É uma das alterações mais importantes para o agravamento da escoliose.
- Meningoceles (6%)
Quando as membranas que revestem a medula (dura máter e aracnoide) apresentam dilatações entre os espaços vertebrais, formando bolsas que contém o líquor. Geralmente localizadas na coluna torácica.
- Dilatação dos forames neurais (40%)
Os nervos que saem e entram na coluna vertebral de ambos os lados passam por orifícios chamados forames intervertebrais. Quando surgem neurofibromas nestes nervos, o crescimento dos neurofibromas vai dilatando os forames lentamente e eles se apresentam alargados na ressonância magnética. Geralmente localizadas na coluna lombar e relacionadas com dor.
Siringomielia (dilatação do canal medular), fraturas e hérnias de disco podem ocorrer nas pessoas com NF1, mas não parecem ser mais frequentes do que na população em geral.
Figura 2 – Alterações na coluna vertebral frequentemente associadas à Neurofibromatose do tipo 1.
Causas
As causas das anormalidades do desenvolvimento ósseo na NF1 ainda estão sendo estudadas, mas algumas possibilidades são:
- Alterações no metabolismo da Vitamina D
- Osteopenia (densidade óssea menor do que o normal)
- Erosão de ossos pela presença dos neurofibromas
- Aumento da pressão dentro da dura máter na medula
- Defeito no desenvolvimento embrionário do folheto mesodérmico por insuficiência da neurofibromina
Ainda não foi possível associar as alterações da coluna vertebral com qualquer tipo de variante genética no gene NF1.
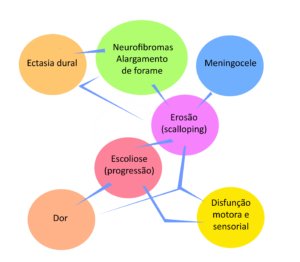
Associação entre as alterações ósseas os sintomas e a progressão da escoliose.
Parece haver uma ligação entre as alterações ósseas, os sintomas e a progressão da escoliose da seguinte maneira: a presença de ectasia dural e de meningocele está associada com neurofibromas nos forames e este conjunto de fatores aumenta a chance de erosões, o que favorece a progressão da escoliose, que causa dor nas costas e redução da função motora e sensorial (Figura 2).
A presença de três ou mais alterações ósseas aumenta significativamente o risco de progressão da escoliose.
Tratamento da escoliose
O tratamento da escoliose requer a avaliação da ortopedia com experiência em NF1 e informações sobre este tema podem ser encontradas em nossa página (ver aqui).
Dr. Lor