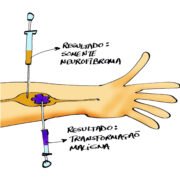
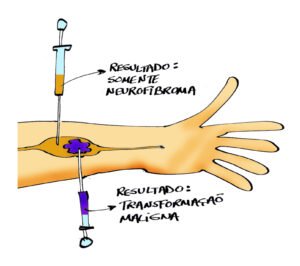
Nas pessoas com neurofibromatose do tipo 1 (NF1), frequentemente precisamos esclarecer se um tumor é um neurofibroma ou se este neurofibroma sofreu uma transformação maligna, o que ocorre em cerca de 5 a 10% dos neurofibromas plexiformes e nodulares.
Essa dúvida ocorre quando uma pessoa com NF1 apresenta um tumor, um neurofibroma plexiforme ou um neurofibroma nodular – já existente, que cresceu rapidamente ou mudou de consistência (ficou mais duro e firme) ou começou a doer (dor forte e persistente) ou atrapalhou a função neurológica ou mais de uma destas mudanças ao mesmo tempo.
Nessa situação acima, temos que agir rapidamente para esclarecer se continua sendo um neurofibroma benigno ou se uma parte dele sofreu transformação para Tumor Maligno da Bainha do Nervo Periférico (TMBNP).
Os TMBNP são tumores agressivos que precisam ser retirados cirurgicamente o mais breve possível, pois não respondem bem à quimioterapia e nem à radioterapia.
Diante dessa dúvida, quais são nossas condutas?
Nos últimos meses, discutimos este problema nas reuniões da equipe do Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais (Dra. Juliana de Souza, Dra. Luíza Rodrigues, Dra. Hérika Martins, Dra. Carolina Feitosa, Enfermeira Marina Corgosinho, Dr. Bruno Cota, Dr. Nilton Rezende, Dr. Renato Viana e Dr. Lor).
Resumimos abaixo algumas das nossas conclusões sobre como saber se um tumor é benigno ou maligno.
- Podemos realizar uma Biópsia
A retirada de uma parte do tumor (por agulha ou por cirurgia) e sua análise no laboratório seria o padrão mais confiável para definirmos se houve a transformação maligna, mas a biópsia apresenta alguns problemas nas pessoas com NF1.
A biópsia pode ser útil em outros tipos de câncer, quando todo o tumor é canceroso. Assim, qualquer fragmento colhido pela agulha poderá mostrar o tumor maligno.
Na NF1 não acontece assim. Nos neurofibromas, a transformação maligna geralmente ocorre em parte do tumor. Então, a biópsia pode colher um fragmento da parte maligna e confirmar o diagnóstico e ajudar a salvar a vida da pessoa.
No entanto, a biópsia por agulha pode pegar apenas um fragmento da parte que continua sendo neurofibroma e não revelar o grande perigo que está ao lado.
Portanto, nas pessoas com NF1, não confiamos na biópsia com agulha quando o resultado é negativo (ver mais informações aqui).
Além disso, para o acompanhamento de neurofibromas ao longo dos anos, biópsias repetidas podem não ser o método mais confortável para as pessoas. Então, precisamos contar com outros métodos que nos ajudem a esclarecer se um neurofibroma sofreu transformação maligna.
- Podemos recorrer aos métodos de diagnóstico por imagens
Em 2022, cientistas chineses (Liu e colaboradores) publicaram uma revisão dos métodos diagnósticos da transformação maligna de neurofibromas, incluindo aqueles disponíveis e outros em estudo, como a Inteligência Artificial (ver aqui o artigo completo, em inglês).
Apesar do número relativamente pequeno de estudos científicos, por causa da NF1 ser uma doença rara, há vantagens e desvantagens de cada método, que serão resumidas a seguir.
Ultrassom
O ultrassom é um método não invasivo e econômico.
Quando um neurofibroma se torna maligno o ultrassom mostra tumores lobulados, com aspecto heterogêneo, com sinais de ampla circulação sanguínea.
No ultrassom de alta resolução, podem ser encontrados sete sinais sugestivos de transformação maligna:
- Tumores maiores do que 5 cm
- Crescimento rápido em semanas ou meses
- Margens irregulares com edema
- Aspecto heterogêneo
- Aspecto infiltrativo
- Vascularização estenótica, oclusiva, trifurcada e arcaica (enquanto nos benignos é hierárquica)
- Contraste aumentado na periferia e inalterado no centro do tumor
No entanto, pelo fato do ultrassom ser um método que exige a presença do médico para ser realizado, seu acesso é mais difícil do que a ressonância magnética ou a tomografia, que podem ser realizados por pessoas com formação técnica. Além disso, a necessidade de realização de ultrassonografia com contraste para identificar a diferença de distribuição dele entre periferia e centro, torna o acesso ao exame ainda mais difícil.
Ressonância Nuclear Magnética (RNM)
A RNM é o método atualmente preferido para identificar tumores malignos, porque apresenta melhor resolução para tecidos moles, como são os neurofibromas.
Na RNM, nas imagens em T1, os neurofibromas benignos e o TMBNP são difíceis de distinguir, mas alguns achados ajudam a definir a possível malignidade:
- Sugestivos de TMBNP:
- Tamanho dos tumores maiores do que 5 cm
- Imagens em T1 com gadolínio com aumento periférico da intensidade no TMBNP (e central nos neurofibromas)
- Presença de hemorragia, necrose e aspecto cístico
- Tumores com margens borradas (“em penugem”) e com edema ao redor
- Sugestivos de neurofibromas benignos:
- Massas bem definidas com alta intensidade em T2
- Presença de área central menos intensa – o “sinal do alvo”
Com estes critérios, alguns estudos sugerem cerca de 65% de sensibilidade e 90% de especificidade para detecção do TMBNP.
A RNM funcional pode aumentar a sensibilidade para 88% e a especificidade para 94% se considerar os índices de difusão.
A RNM em 3 dimensões pode auxiliar na detecção precoce de aumentos volumétricos dos tumores e a arga tumoral aumentada na ressonância de corpo inteiro também pode sugerir a possibilidade de transformação maligna.
Limitações da RNM são:
- É um método dependente da subjetividade do examinador
- Há poucos estudos com os neurofibromas “atípicos” (tumores com padrão intermediário entre benigno e maligno)
Tomografia computadorizada (TC)
Não dispomos ainda de critérios seguros para distinguir neurofibromas de TMBNP apenas com a TC. Alguns estudos estão associando a TC com aprendizado de máquina (inteligência artificial) e podem no futuro trazer uma contribuição a esta questão.
Novamente, temos que observar que quanto à ressonância magnética e tomografia, uma vez que para alguns dos critérios estabelecidos para a distinção do tumor maligno é necessária a administração de contraste, é necessário que um médico esteja presente.
Positron Emission Tomography (PET)
O método mais popular combina o PET com a CT (PET/CT), usando o contraste radioativo 18F-FDG.
Os achados mais importantes dos TMBNP no PET/CT são:
- Aumento da captação do contraste (SUV maior que 3,15)
- Heterogeneidade da captação
Limitações do 18FDG-PET/CT são:
- Sobreposição de SUV entre neurofibromas atípicos e TMBNP
- Sensibilidade 100% lesões assintomáticas – especificidade para negativo 100% – especificidade para positivo 46%
- Custo elevado
Alguns estudos estão sendo realizados com novos marcadores, como 68Ga-PSMA PET/CT e 11-Metionina, mas ainda não temos um consenso sobre seu uso nas pessoas com NF1.
Combinação de PET com RNM (PET/RNM)
Ainda com poucos estudos em NF1 e sem disponibilidade em nosso meio.
Radiogenômica
Trata-se de novos métodos em estudo, que procuram associar os resultados das imagens com análise genética. Alguns dos autores do estudo (Liu e outros) dividiram o gene NF1 em 5 domínios e associaram com dados clínicos e imagens de RNM e encontraram forte correlação com a transformação maligna.
Conclusões
Ainda não temos um consenso internacional, mas o indicador mais forte de transformação maligna é o aumento significativo do tamanho de um neurofibroma.
Nosso desafio futuro será encontrar correlações entre imagens e os 6 tipos histológicos de tumores mais comuns na NF1:
- Neurofibroma
- Neurofibroma com atipias celulares
- Neurofibroma celular
- ANNUBP – neurofibromas atípicos de potencial biológico incerto
- TMBNP de baixo grau
- TMBNP de alto grau
Por enquanto, precisamos verificar em nosso meio a viabilidade do ultrassom contrastado e a disponibilidade de ultrassonografistas com expertise nesse tipo de exame, para que possamos considerá-lo na nossa rotina de cuidados. Além disso, no fututo, combinar métodos e avançar nos estudos com inteligência artificial.
Belo Horizonte, 13 de julho de 2023




 Participaram da banca examinadora os professores (na foto, da esquerda para a direita) Maria Raquel Santos Carvalho (UFMG), Juliana Ferreira de Souza (do Centro de Referência em Neurofibromatoses e do Centro Universitário UNIBH), Marcelo Rizzatti Luizon (UFMG), Wagner Carlos Santos Magalhães (do Instituto Mário Penna) e Renan Pedra de Souza (UFMG).
Participaram da banca examinadora os professores (na foto, da esquerda para a direita) Maria Raquel Santos Carvalho (UFMG), Juliana Ferreira de Souza (do Centro de Referência em Neurofibromatoses e do Centro Universitário UNIBH), Marcelo Rizzatti Luizon (UFMG), Wagner Carlos Santos Magalhães (do Instituto Mário Penna) e Renan Pedra de Souza (UFMG). Segundo, porque vi a dedicação da Cinthia com o projeto e seu desejo de continuar estudando as neurofibromatoses no seu regresso para a Universidade Federal da Bahia, dando-me a impressão de que seu contato conosco frutificará em novas pesquisas importantes. Além disso, seus orientadores e os demais professores da banca mostraram grande interesse pelo tema, sinalizando que novos alunos de pós-graduação interessados em problemas das neurofibromatoses podem surgir no campo da genética, dando continuidade a esta linha de pesquisa.
Segundo, porque vi a dedicação da Cinthia com o projeto e seu desejo de continuar estudando as neurofibromatoses no seu regresso para a Universidade Federal da Bahia, dando-me a impressão de que seu contato conosco frutificará em novas pesquisas importantes. Além disso, seus orientadores e os demais professores da banca mostraram grande interesse pelo tema, sinalizando que novos alunos de pós-graduação interessados em problemas das neurofibromatoses podem surgir no campo da genética, dando continuidade a esta linha de pesquisa.