O que são as “NEUROFIBROMATOSES”?
Algumas informações iniciais e úteis sobre doenças genéticas
O termo Neurofibromatose vem da combinação de duas palavras em medicina: neuro e fibroma. Neuro significa relacionado aos nervos e fibroma é um crescimento exagerado (ou tumor) de células parecidas com fibras.
Um neurofibroma, portanto, é um tumor causado pelo crescimento de células relacionadas com os nervos.
As “neurofibromatoses” são doenças genéticas causadas por mudanças (variantes patogênicas – antes chamadas de mutações) em alguns genes, que acontecem por acaso quando as células se multiplicam.
Genes são as estruturas que controlam nosso crescimento e desenvolvimento e determinam nossas características biológicas, como a cor do cabelo, altura, nosso tipo sanguíneo, etc.
Uma variante patogênica pode alterar o funcionamento do gene, modificando a proteína que ele produz e assim alterar as atividades das células e dos órgãos.
A variante patogênica num gene pode estar presente no espermatozoide ou no óvulo e dessa união resultar uma criança com uma doença genética. O bebê que nasce com a variante patogênica, por exemplo, nas “neurofibromatoses”, irá apresentar deficiência de alguma proteína que seja importante para o desenvolvimento normal do sistema nervoso, da pele e dos ossos. Assim, o bebê poderá apresentar a doença que se manifesta no desenvolvimento inadequado desses órgãos.
Se nenhum dos pais tem uma das “neurofibromatoses”, a descoberta em um dos filhos (ou filhas) é chamada de variante patogênica nova na família, ou seja, uma alteração que aconteceu por acaso quando o óvulo da mãe ou o espermatozoide do pai foram formados.
Embora muitos pais fiquem preocupados achando que fizeram algo de errado para causar esta variante patogênica, sabemos que isto não é verdade. Até agora desconhecemos qualquer coisa que os pais ou mães poderiam ter feito para causar qualquer uma destas doenças.
A partir do nascimento de uma pessoa com a variante nova, essa pessoa carrega a tal variante em metade dos seus espermatozoides ou de seus óvulos, então ela tem a chance de passar para seus filhos ou filhas a mesma variante, por isso dizemos que a doença se torna hereditária.
Em algumas doenças genéticas, como nas “neurofibromatoses”, basta um dos genes (de origem paterna ou materna) possuir a variante patogênica para a doença se manifestar no novo indivíduo (de ambos os sexos). Por isso dizemos que as “neurofibromatoses” se transmitem de forma dominante. Isto quer dizer que um pai ou uma mãe com a NF1, por exemplo, tem 50% de chance de ter uma filho ou filha com a mesma doença em cada gestação.
Em certas doenças genéticas quem tem a variante patogênica em seus genes sempre apresenta a doença a partir de algum momento, ou seja, ela tem a expressão completa. Isto quer dizer que uma doença genética com expressão completa não fica “escondida”, não salta gerações. Por exemplo, não há risco de um neto herdar a NF1 de um avô se o pai ou a mãe não apresentarem também sinais da doença.
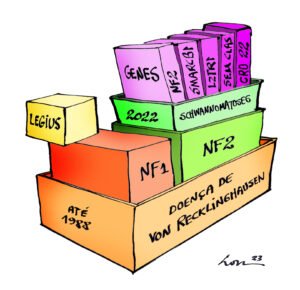
As “Neurofibromatoses”
Existem cerca de 80 mil brasileiras e brasileiros com algumas doenças genéticas que eram agrupadas sob o nome de Doença de von Recklinghausen.
Nos últimos 40 anos, à medida que o conhecimento científico foi aumentando, foram descobertas duas doenças genéticas diferentes onde se pensava haver apenas a Doença de von Recklinghausen. Passamos então a diagnosticar a Neurofibromatose do tipo 1 (NF1) e a Neurofibromatose do Tipo 2 (NF2).
Depois, foi identificada a Síndrome de Legius, uma doença que é parecida com a NF1 (porque a Legius apresenta manchas café com leite, efélides e dificuldades cognitivas, mas não apresenta tumores), e que era diagnosticada equivocadamente como NF1.
Em seguida, descobriu-se outra doença que se confundia com a NF2, que foi denominada Schwannomatose.
Logo foram encontrados dois genes diferentes (SMARCB1 e LZTR1) responsáveis pela Schwannomatose.
Portanto, foi ficando cada vez mais difícil para as pessoas – que não são especialistas – compreender este grupo de doenças. Assim, foi criada uma comissão de especialistas para organizar melhor as “neurofibromatoses”.
NOVOS NOMES PARA AS DOENÇAS ANTES CHAMADAS DE “NEUROFIBROMATOSES”
Depois de um trabalho de três anos, realizado por uma equipe internacional de especialistas, foram definidos novos nomes para as doenças que antes eram agrupadas como Neurofibromatoses.
Nosso Centro de Referência participou da elaboração dos novos critérios e se você desejar conhecer o artigo original (em inglês) que definiu a mudança e os critérios diagnósticos para estas doenças basta clicar aqui.
Elas têm algumas manifestações em comum, por isso estiveram juntas por muitos anos (ver ilustração acima).
O que elas têm em comum?
Elas são doenças raras, que possuem em comum as manchas cutâneas cor de café com leite, a formação de tumores múltiplos derivados do sistema nervoso.
À medida que a ciência aumentou nosso conhecimento, hoje somos capazes de reconhecer pelo menos 6 doenças diferentes.
NF1 – Neurofibromatose do tipo 1 (NF1)
SWN-NF2 – Schwannomatose relacionada ao gene NF2 (antes chamada de Neurofibromatose do tipo 2 ou NF2)
SWN-SMARCB1 – Schwannomatose relacionada ao gene SMARCB1 (antes chamada de Schwannomatose)
SWN-LZTR1 – Schwannomatose relacionada ao gene LZTR1 (antes também chamada de Schwannomatose)
SWN-NOS – Schwannomatose com diagnóstico clínico, mas sem análise genética
SWN-CRO 22 – Schwannomatose relacionada a alterações no cromossomo 22, mas sem alterações nos genes SMARCB1 e LZTR1.
SWN-NEC – Schwannomatose com diagnóstico clínico, mas cuja análise genética não conseguiu revelar variantes patogênicas.
Como fazer o diagnóstico destas doenças?
O diagnóstico e a orientação clínica da NF1 podem ser encontrados em português neste site (clicando aqui) ou no artigo original em inglês clicando aqui .
Ambos os diagnósticos e orientações da NF1 e da SR-NF2 podem ser realizados com exames clínicos e exames de imagem, sem necessariamente a análise genética.
As demais doenças (Legius e Schwannomatoses) precisam da análise genética para serem corretamente identificadas e as pessoas portadoras e suas famílias serem devidamente orientadas.
Os critérios diagnósticos para cada uma delas podem ser encontrados neste mesmo site, e para isto basta entrar no menu de cada uma delas.
Quadro 1 – Principais características da Neurofibromatose do tipo 1 (NF1) e das Schwannomatoses (SWN).
| Doenças | NF1 | Schwannomatoses (SWN) | |||||
| NF1 | SWN-NF2 | SWN-SMARCB1 | SWN –LZTR1 | SWN- NEC sem variante | SWN-CRO22 ligada ao Crh 22 | SWN-NOS sem análise do DNA | |
| Gene
(autossômicos) |
NF1 | NF2 | SMARCB1 | LZTR1 (*) | ? | Crh 22 | ? |
| Penetrância Expressão
De novo |
100%
completa 50% |
100%
variável 60% |
70%
mosaico 30% |
70%
mosaico 30% |
DIPE | DIPE | DIPE |
| Início dos sintomas | Infância | Segunda década | Depois dos 30 | Depois dos 30 | Depois dos 30 | Depois dos 30 | Depois dos 30 |
| QP mais comum | MCL | Baixa audição | Dor | Dor | Dor | Dor | Dor |
| MCL | 98% | 2% | raras | raras | DIPE | DIPE | DIPE |
| Dor neuropática | 20% | Rara | Sim | Sim | DIPE | DIPE | DIPE |
| Alterações Audição | DPA | perda | não | não | não | não | não |
| Efélides | sim | não | não | não | não | não | não |
| Tumores | nf
plexif glio feo TMBNP |
sch vest
sch derm menin ependi |
sch perif
(vest? menin?) |
sch perif | sch perif | sch perif | sch perif |
| Alterações oculares | Lisch
coroide |
opacid len
hamart memb epi |
não | não | DIPE | DIPE | DIPE |
| Alterações cognitivas | 70% | não | não | não | DIPE | ? | DIPE |
| Alterações ósseas | esf
ossos longos |
hiperostose | não | não | DIPE | DIPE | DIPE |
Legenda: DIPE: Dados insuficientes para estatísticas – a SWN sem análise genética, a SWN por alterações no cromossomo 22 e a SWN sem variantes patogênicas identificadas são mais raras e foram recentemente classificadas; MCL: manchas café com leite; DPA: desordem do processamento auditivo; tipos de tumores: nf: neurofibroma; plexif: neurofibroma plexiforme; glio: glioma; feo: feocromocitoma; TMBNP: tumor maligno da bainha do nervo periférico; sch vest: schwannoma vestibular; sch derm: schwannoma intradermal; menin: meningioma; ependi: ependimoma; sch perif: schwannoma periférico; Lisch: nódulos de Lisch; coroide: alterações da coroide; opacid len: opacidade lenticular juvenil; hamart: hamartoma; memb epi: membrana epirretiniana; esf: displasia do esfenoide; ossos longos: displasia da tíbia e ossos longos; (*) o gene LZTR1 pode estar envolvido na Síndrome de Noonan.
Observações importantes
Como elas são consideradas doenças raras, é razoável esperar que a maioria dos profissionais da saúde tenha alguma dificuldade em diagnosticá-las e tratá-las de forma adequada. Sabendo que existem cerca de 5 mil doenças raras distribuídas em 3 a 5% da população mundial, fica evidente que é difícil para qualquer profissional da saúde conhecer cada uma delas.
Além disso, algumas situações históricas contribuíram para a construção de imagens equivocadas sobre estas doenças, como o próprio agrupamento de três doenças muito diferentes sob o mesmo nome (neurofibromatoses), assim como confundi-las com outras doenças raras, como a Síndrome de Proteus, que se tornou famosa por causa de Joseph Merrick, o chamado Homem Elefante.
A raridade, a complexidade e a variabilidade clínica destas quatro doenças fazem com que diante de uma pessoa com suspeita de uma delas os profissionais de saúde identifiquem facilmente as lesões cutâneas, mas em geral desconheçam que elas são a superfície do problema, o que pode dificultar a conduta clínica correta.
O diagnóstico da NF1 é geralmente simples e fácil de ser alcançado no exame clínico ambulatorial. Por outro lado, o diagnóstico e orientação para as Schwannomatoses exigem exames complementares e experiência do profissional de saúde.
Em seguida ao diagnóstico, no entanto, é preciso reconhecer a complexidade das condutas necessárias para se evitar as complicações, aumentar a qualidade e expectativa média de vida.
Nosso site pretende ser compreensível para todos os profissionais da saúde, especialistas ou não, assim como para as famílias e pessoas com estas doenças , porque sabemos que a principal alternativa no longo prazo para as doenças raras é o fortalecimento progressivo do conhecimento dos próprios doentes e suas famílias sobre a doença que os acomete.
O que geralmente motiva a consulta
As manchas cutâneas cor de café com leite (MCL) e tumores cutâneos geralmente fazem parte da queixa principal e constituem elementos fundamentais para o diagnóstico destas quatro doenças. No entanto, outros sinais e sintomas, assim como complicações são extremamente diferentes quanto às características e momento de aparecimento.
Na NF1, o diagnóstico demora cerca de 3 anos para ser estabelecido e as principais queixas ocorrem geralmente na infância, e são as seguintes em ordem de frequência:
- Quase todos apresentam uma ou mais dificuldades cognitivas (para aprender a andar e falar, problemas no desempenho escolar e baixa habilidade nas relações sociais, nas atividades esportivas e musicais);
- Quase todos apresentam um ou mais problemas estéticos que resultam em discriminação social (macrocrania, neurofibromas cutâneos, deformidades corporais provocadas por neurofibromas plexiformes congênitos);
- A maioria apresenta cefaleia crônica, baixa estatura e baixo peso.
- Cerca de um quarto das pessoas com NF1 apresenta tumores internos (englobando-se aqui desde os gliomas ópticos, ou em outras partes do sistema nervoso central, até neurofibromas plexiformes em crescimento ou em processo de transformação maligna para neurofibrossarcomas);
- Um em cada cinco apresenta hipertensão arterial secundária a displasias da artéria renal, ou feocromocitoma ou a mecanismo ainda não conhecido.
- Um em cada dez apresenta deformidades esqueléticas (desde a escoliose benigna até graves deformidades distróficas da coluna vertebral, passando pela displasia dos ossos longos, especialmente da tíbia, com ou sem pseudoartrose, até a displasia da asa menor do osso esfenoide com proptose ocular).
Na Schwannomatose relacionada ao gene NF2, o diagnóstico demora em média 8 anos para ser estabelecido e as principais queixas acontecem no início da vida adulta e são zumbido e diminuição da audição, o que faz com que o diagnóstico inicial seja feito pela otorrinolaringologia ou pela neurologia.
Nas Schwannomatoses relacionadas aos genes SMARCB1 e LZTR1, que geralmente se manifestam depois dos 30 anos, o diagnóstico demora cerca de 12 anos para ser realizado e a queixa principal é dor, de caráter neuropático, em um ou mais dos schwannomas cutâneos ou profundos, e o diagnóstico geralmente é feito pelos neurologistas.
Ver nas páginas seguintes deste site como identificar cada uma destas doenças e veja também as cartilhas que elaboramos para facilitar sua compreensão.
Nosso trabalho depende da sua generosidade
Se você deseja fazer uma doação, a conta da AMANF é:
BANCO CREDICON
Nome: AMANF ASSOCIAÇÃO MINEIRA DE APOIO AOS PORTADORES D
Banco:756
Agência: 4027
Conta Corrente: 20261001 dígito 2
CNPJ: 05.629.505/0001-33
PIX +55 (31) 99971-0622