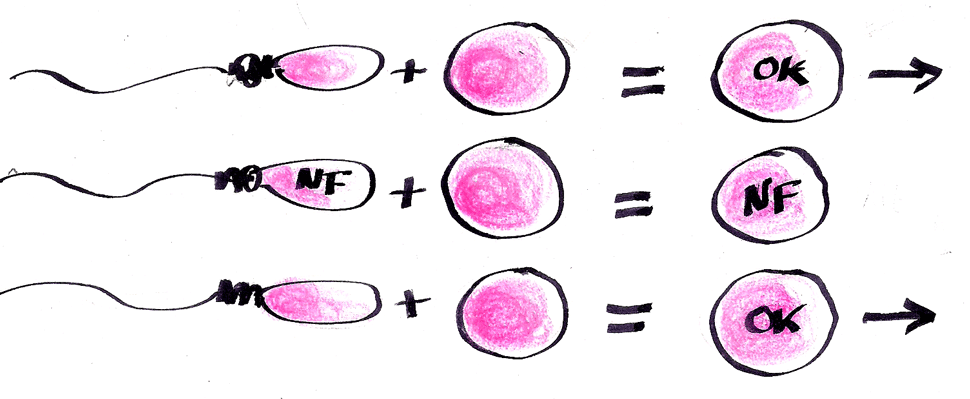

“Meu filho tem 5 anos e somente manchas café com leite, são mais de 6, mas o teste genético foi negativo para NF1. Como devo agir? ”. MVD, de São José dos Campos, SP.
Cara M, obrigado pela sua excelente pergunta que pode ser útil a muitas famílias na mesma situação.
Quando uma criança apresenta dois ou mais critérios para o diagnóstico de neurofibromatose do tipo 1 (NF1), temos praticamente certeza de que ela possui a doença. Por exemplo, 6 ou mais manchas café com leite e sardas nas axilas (efélides), ou 6 ou mais manchas café com leite e Nódulos de Lisch na íris, ou 6 ou mais manchas café com leite e um neurofibroma plexiforme.
No entanto, quando a criança apresenta apenas as manchas café com leite, precisamos examinar com bastante cuidado as características das manchas, para sabermos se são as manchas tipicamente encontradas na NF1 ou se são manchas atípicas, ou seja, não estão associadas à NF1.
(Ver no final um comentário da Dra. Luciana Baptista Pereira sobre estes termos e a versão em inglês que foi aprimorada pelo Dr. Vincent Riccardi e pelo Dr. Nikolas Mata-Machado e a publicação no site da NFMidwest)
Primeira pergunta: como são as manchas café com leite?
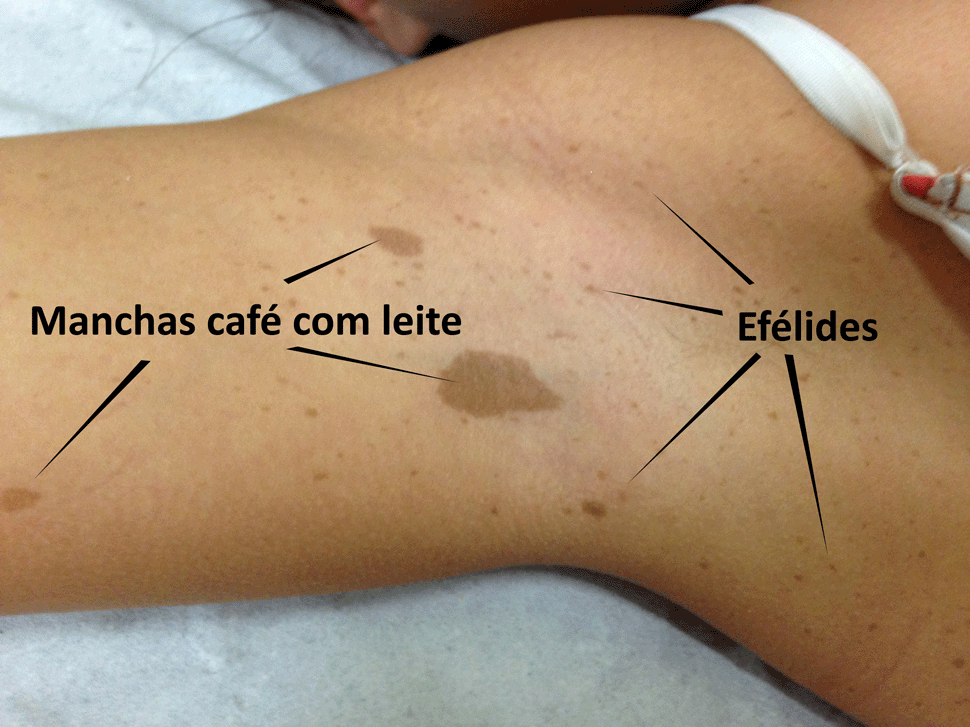
As manchas típicas da NF1 são ovaladas, de limites bem definidos, de cor café com leite homogênea e se localizam principalmente no tronco (ver figura), formando ilhas isoladas. As manchas atípicas, que não estão obrigatoriamente associadas à NF1, geralmente têm formatos muito variáveis, seus limites são pouco definidos e podem ser encontradas em qualquer parte do corpo, formando ilhas de contornos irregulares (ver figura).
A segunda questão é o tamanho das manchas. O critério diagnóstico para NF1 exige que as manchas café com leite típicas tenham mais de meio centímetro na infância e mais de 1 cm a partir da adolescência. Então, é preciso distinguir manchas café com leite das efélides, que são sardas localizadas em locais onde não bate sol, como as axilas e a as virilhas (ver figura) e que são o segundo critério para o diagnóstico de NF1.
Segunda pergunta: quantas são as manchas café com leite que são típicas?
Há uma relação direta entre o número de manchas típicas no primeiro ano de vida e a chance de aparecimento de outros sinais da NF1 nos anos seguintes, como mostrou um estudo científico realizado por Nunley e colaboradores e publicado em 2009 (ver AQUI ). Os pesquisadores verificaram que uma criança com 6 manchas típicas no primeiro ano de vida tem 40% de chance de apresentar outros sinais da NF1 nos próximos cinco anos. Mas uma criança com 9 ou mais manchas típicas no primeiro ano de vida tem a chance de 100% de apresentar outro sinal da NF1 nos próximos 5 anos de vida.
Em outras palavras, grande parte (75%) das crianças com 6 ou mais manchas café com leite típicas terá seu diagnóstico de NF1 confirmado até os 6 anos de idade e para a imensa maioria (92%) a NF1 será confirmada até os 10 anos de idade.
Por outro lado, nenhuma das crianças apresentou diagnóstico de NF1 quando elas tinham apenas 1 até 5 manchas café com leite durante o período do estudo realizado. Além disso, somente 5% das crianças com manchas atípicas apresentou NF1 ao longo do estudo.
Em resumo, até 3 manchas típicas, provavelmente não há doença associada; de 3 a 5 manchas, pensar em outras doenças; 6 ou mais manchas, agir como se fosse NF1.
Terceira pergunta: devemos fazer o teste genético numa criança pequena quando ela só apresenta só e exclusivamente as típicas manchas café com leite?
Esta é uma questão frequente em nosso Centro de Referência, porque todos nós gostaríamos de saber imediatamente o diagnóstico definitivo para podermos agir de acordo. No entanto, sempre avalio com os pais o seguinte: o resultado do teste genético vai mudar nossa conduta?
Se o teste vier positivo para NF1, nossa conduta será observar a criança e reavaliarmos clinicamente a cada ano se não houver novidades.
Se o resultado do teste genético para NF1 vier negativo[1], nossa conduta será observar a criança, que deve levar uma vida absolutamente normal e reavaliarmos clinicamente a cada ano se não houver novidades.
Ora, neste aspecto o teste não mudaria nossa conduta.
Por outro lado, se o teste vier negativo temos que pensar em outras doenças que podem também causar manchas café com leite (embora na maioria destas outras doenças as manchas geralmente sejam atípicas). Por exemplo, Síndrome de Legius, Osteodistrofia de Albright e Mastocitose.
É bom lembrarmos que algumas formas de NF1 apresentam poucas manchas café com leite.
Chegamos então na sua pergunta, cara M: quais as outras condições que podem causar manchas café com leite?
Primeiro, existem as manchas café com leite isoladas que são encontradas em 10% das pessoas sadias. Também há uma forma de mancha café com leite familial, em que vários membros de uma família apresentam uma mancha, sem que isto seja sinal de doença.
Por outro lado, há um grupo relativamente grande de doenças que podem causar manchas café com leite, geralmente menos de 6 manchas e atípicas: Neurofibromatose do tipo 2, Schwannomatose, Síndrome de Legius, Síndrome de Noonan, Rasopatias, Síndrome de deficiência de reparo do DNA, Piebaldismo, Síndrome de Bloom e Anemia de Fanconi, Mastocitose e Osteodistrofia de Albright.
A diferenciação entre cada uma destas doenças requer um profissional com experiência clínica em NF1 e em várias doenças raras e um resumo de suas características pode ser encontrado em nosso artigo publicado em 2013 (ver AQUI ).
Um comentário importante: manchas típicas e atípicas são termos adequados?
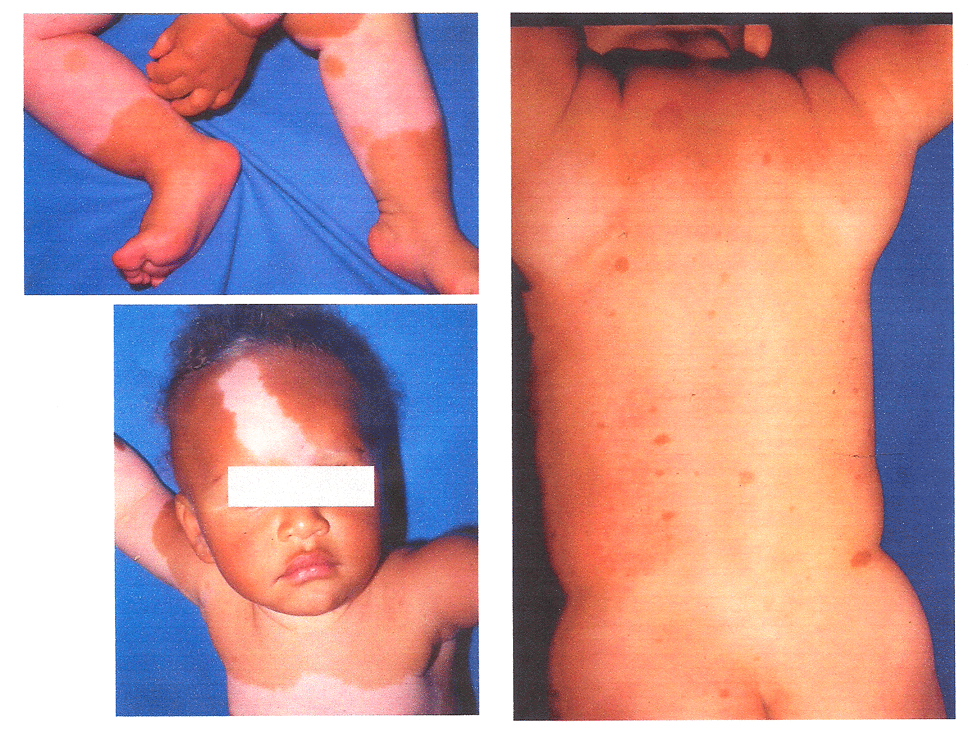
A Professora Dra. Luciana Baptista Pereira diz o seguinte: “Discordo em separar as manchas café com leite em atípicas e típicas. Separaria em manchas café com leite (costa da Califórnia) e nevo hiperpigmentado (costa da Flórida). Há várias síndromes com manchas café com leite da mesma forma que as da NF1, para mim idênticas e impossíveis de separar pela mancha em si. Veja abaixo a foto de uma criança com piebaldismo como exemplo”.
English version
Improved by Dr. Vincent Riccardi and Dr. Nikolas Mata-Machado.
When a child has two or more criteria for the diagnosis of neurofibromatosis type 1 (NF1), we are almost certain that she has the disease. For example, 6 or more cafe au lait spots (CALMS) and freckles in the axilas, or 6 or more CALMS and Lisch Nodes on their iris, or 6 or CALMS and a plexiform neurofibroma.
However, when the child has only CALMS, we need to examine carefully the characteristics of the spots to find out if they are the spots typically found in NF1 or if they are atypical spots, that is, they are not associated with NF1.
First question: How to characterize the Café-au-lait macule (CALM)?
Typical NF1 CALMS are oval, with well-defined limits, homogeneous color similar to café au lait and located mainly in the trunk (see figure), forming isolated islands. Atypical spots, which are not ordinary for NF1, usually have very variable shapes, their boundaries are poorly defined and can be found anywhere on the body, forming irregularly contoured islands (see figure).
The second question is the size of the spots. The diagnostic criterion for NF1 requires that typical CALMS be more than half a centimeter in childhood and more than 1 cm from adolescence. Therefore, it is necessary to distinguish CALMS from the freckles, which are located in places where the sun does not touch, such as the armpits and groin (see figure) and they are a second criterion for the diagnosis of NF1.
Second question: How many CALMs are typical?
There is a direct relationship between the number of typical spots in the first year of life and the chance of other NF1 signs appearing in subsequent years, as shown in a scientific study by Nunley and colleagues published in 2009 (see here www.jamanetwork.com/journals/jamadermatology/fullarticle/712161 ). The researchers found that a child with 6 typical spots in the first year of life has a 40% chance of presenting other NF1 signs within the next five years. However, a child with 9 or more typical spots in the first year of life has a 100% chance of having another NF1 sign in the next 5 years of life.
In other words, a large proportion (75%) of children with 6 or more typical CALMS will have their diagnosis of NF1 confirmed by age 6 and for the vast majority (92%) NF1 will be confirmed by age 10 of age.
On the other hand, none of the children presented a diagnosis of NF1 when they had only 1 to 5 CALMS during the study period. In addition, only 5% of the children with atypical spots had NF1 throughout the study.
In summary, up to 3 typical spots, there is probably no associated disease; 3 to 5 spots, we may think of other diseases; 6 or more stains, act as if it were NF1.
Third question: should we do the genetic test in a small child when it only presents only CALMS?
This is a frequent question in our Reference Center because we would all like to know immediately the definitive diagnosis so that we can act accordingly. However, I always evaluate with parents the following: Will the genetic test result change our behavior?
If the test is positive for NF1, our behavior will be to observe the child, and observe for other NF1 elements as the child ages.
If the result of the genetic test for NF1 is negative, our behavior will be to observe the child, who must lead a normal life and reassess clinically each year if there is no news.
Now in this respect the test would not change our conduct.
On the other hand, if the test comes negative we have to think about other diseases that can also cause brown spots with milk (although in most of these other diseases the spots are usually atypical). Legius syndrome, Albright Osteodystrophy, Mastocytosis. Also, some forms of NF1 have very few CALMs (Huson, et al.)
Summary: So we came to your question: what other conditions that can cause CALMS?
First, there are the isolated CALMS that are found in 10% of healthy people. There is also a form of CALM in which several members of a family have a spot, without this being a sign of disease. Finally, it is possible that parent great skin color differences could lead to atypical CALM in their children.
On the other hand, there is a relatively large group of diseases that can cause CALM, usually less than 6 spots and atypical: Neurofibromatosis type 2, Schwannomatosis, Legius Syndrome, Noonan Syndrome, Albright Osteodystrophy.
The differentiation between each of these diseases requires a professional with clinical experience in NF1 and various rare diseases and a summary of their characteristics can be found in our article published in 2013 (see here https://www.ncbi.nlm.nih.gov/pubmed/24676443 ).
[1] Neste caso, é preciso que o laboratório se certifique de que não houve a deleção do gene, o que poderia causar um resultado falso negativo na análise do gene NF1.
Cafe Au Lait Spots and Diagnosis of NF1
Typical NF1 CALMS are oval, with well-defined limits and uniform color similar to “coffee with milk” and located mainly in the trunk, forming isolated islands. Atypical spots, which are not ordinary for NF1, usually have very variable shapes, their boundaries are poorly defined and can be found anywhere on the body, forming irregularly contoured islands (see figure).
There is a direct relationship between the number of typical spots in the first year of life and the chance of other NF1 signs appearing in subsequent years, as shown in a scientific study by Nunley and colleagues published in 2009 (see here www.jamanetwork.com/journals/jamadermatology/fullarticle/712161 ). The researchers found that a child with 6 typical spots in the first year of life has a 40% chance of presenting other NF1 signs within the next five years. However, a child with 9 or more typical spots in the first year of life has a 100% chance of having another NF1 sign in the next 5 years of life.