Hoje, responderei a duas perguntas diferentes, mas que têm uma resposta em comum.
“Pelo que tenho lido no seu blog, você defende que as manchas café-com-leite, típicas da NF1, ou já nascem com a criança ou aparecem daí a uns meses, aumentando de número, mas chegando até a desaparecer na idade adulta. No entanto, da minha participação em fóruns, etc, ouço vários relatos de pessoas afectadas dizendo que lhe estão a aparecer mais e mais manchas café com leite. O que diz desta situação?” AG, de Portugal.
“Gostaria de saber se a quantidade de manchas e pintas café com leite tem relação com o grau da NF. E gostaria de saber se existe realmente esses graus na síndrome? Meu filho tem 8 meses e diariamente as manchas e pintas aumentam. Já fizemos o mapeamento é realmente ele é portador da síndrome, ele é mutação já que ninguém tem a NF. Fiz a Ressonância que não apresentou problemas, mas meu filho está atrasado em relação a sentar, rolar e engatinhar..”. K, de local não identificado.
Prezadas AG e K, obrigado pela participação no blog.
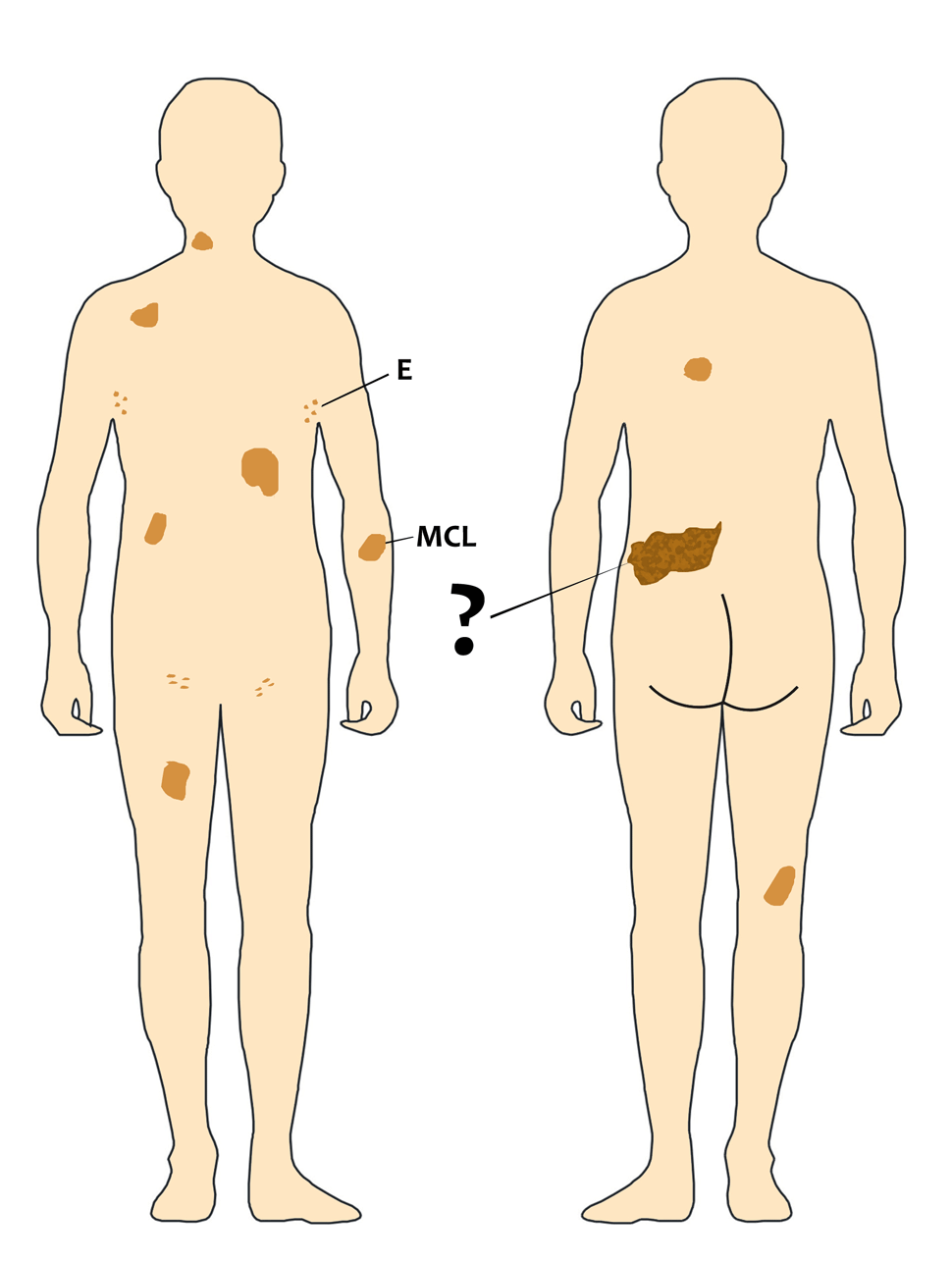
De fato, muitas pessoas relatam o aumento do número de supostas manchas café com leite (MCL) porque geralmente estão se referindo às efélides (E), ou seja, aquelas manchas menores (pintas ou sardas) que estas, sim, na minha experiência, continuam a aumentar ao longo da vida.
Por outro lado, as verdadeiras MCL são congênitas, isto é, estão presentes ao nascimento ou ficam evidentes nas primeiras semanas de vida assim que a criança entra em contado com a luz solar direta ou indiretamente. Minha impressão clínica é de que as MCL não aumentam de número durante a vida e apenas acompanham, proporcionalmente, o crescimento das pessoas. Ao longo da vida adulta e nos idosos as MCL começam a diminuir em pigmentação, ou seja, podem ficar mais claras ao longo dos anos.
Sabemos que a mutação (ou deleção) no gene NF1 é a causa das manchas café com leite e das efélides, mas não sabemos o porquê elas aparecem em certa quantidade e em determinado local. Curiosamente, o aspecto anatômico das MCL é idêntico ao das efélides, fazendo com que alguns estudiosos proponham que sejam considerados um critério único. Do meu ponto de vista, ainda que as MCL e efélides apresentem a mesma histologia, o momento de seu aparecimento (ao longo da infância) e sua evolução (podem continuar a aumentar durante a vida) sugerem que sejam mecanismos genéticos diferentes.
A outra questão é quanto ao significado clínico das MCL e efélides para definirmos a gravidade da doença, o que imagino que seja aquilo que a leitora K. chamou de grau. Até o presente momento, não conheço qualquer evidência científica de que a quantidade e tamanho das MCL e efélides tenham qualquer relação com a gravidade da NF1. Ou seja, a gravidade da NF1 não depende do número, idade de aparecimento e tamanho das MCL e efélides.
Quem desejar saber mais sobre como classificamos a gravidade da NF1 em mínima, leve, moderada ou grave, por favor veja AQUI).