Foi publicado na última semana (30/3/2017) na revista científica internacional American Journal of Medical Genetics Part A o nosso artigo intitulado “Bilateral second toe superposition associated with neurofibromatosis type 1 and NF1 whole gene deletion” (tradução: Superposição bilateral do segundo dedo dos pés está associada à deleção completa do gene NF1 – ver a referência do artigo em inglês AQUI)
Esta publicação numa revista de alta qualidade científica nos traz mais segurança para orientarmos as famílias com este sinal (ver a figura), cerca de 10% das famílias com NF1, no sentido de procurarmos a confirmação da deleção por meio de teste genético (MPLA), uma vez que esta forma da NF1 requer alguns cuidados especiais.
(Ver aqui AQUI excelente revisão científica recente, de 2017, sobre os aspectos clínicos e genéticos das deleções do gene NF1).
Este estudo científico foi realizado pelos pesquisadores do Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais (Luiz Rodrigues, Juliana Souza, Luíza Rodrigues e Nilton Rezende), em colaboração com pesquisadores do Laboratório Hermes Pardini (Frederico Malta, Juliana Rodrigues e Patricia Couto), sob a orientação do médico pioneiro das Neurofibromatoses o Dr. Vincent Riccardi, do The Neurofibromatosis Institute, La Crescenta, Califórnia, Estados Unidos.
Em resumo, a American Journal of Medical Genetics Part A publicou as informações a seguir.
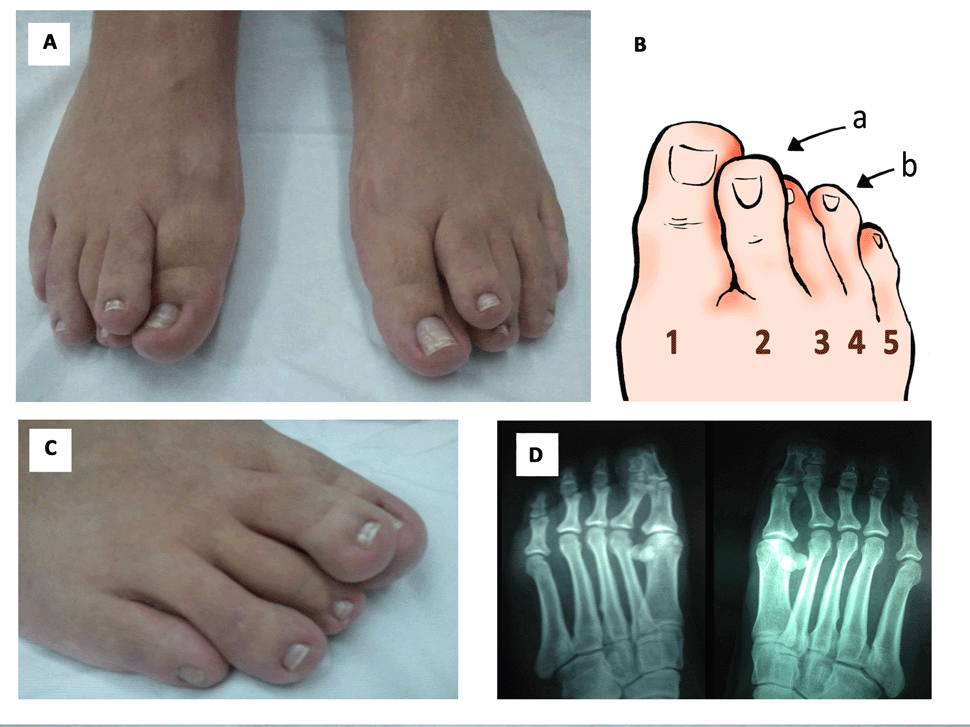
Em alguns indivíduos atendidos no CRNF com o diagnóstico de NF1 notamos a superposição do segundo dedo sobre o terceiro de ambos os pés. Chamamos a este sinal anatômico de Superposição do Segundo Dedo (em inglês a sigla ficou STS) (outras imagens do STS depois do texto abaixo).
Numa etapa seguinte, nós perguntamos por meio de um questionário especial a 445 pessoas com NF1 atendidas e diagnosticadas no nosso CRNF quem possuía o STS. Entre as 162 que responderam ao questionário, 11,9% delas confirmaram a presença da superposição do segundo dedo.
Baseados nestas observações anteriores, nós fotografamos os pés de 187 pessoas COM NF1 e 194 SEM NF1 e as fotografias foram avaliadas por 3 examinadores independentes, que não sabiam quem tinha ou não NF1. O resultado mostrou que o STS estava presente em 8% das pessoas com NF1, ausente em 80,2% e indeterminado em 11,8% delas. Na população controle (isto é, sem NF1), o STS estava presente somente em 0,5%, ausente em 95,4% e indeterminado em 4,1% delas. Ou seja, o STS foi 16 vezes mais presente na NF1 do que nas pessoas sem NF1, o que foi estatisticamente significativo (P<0,0001).
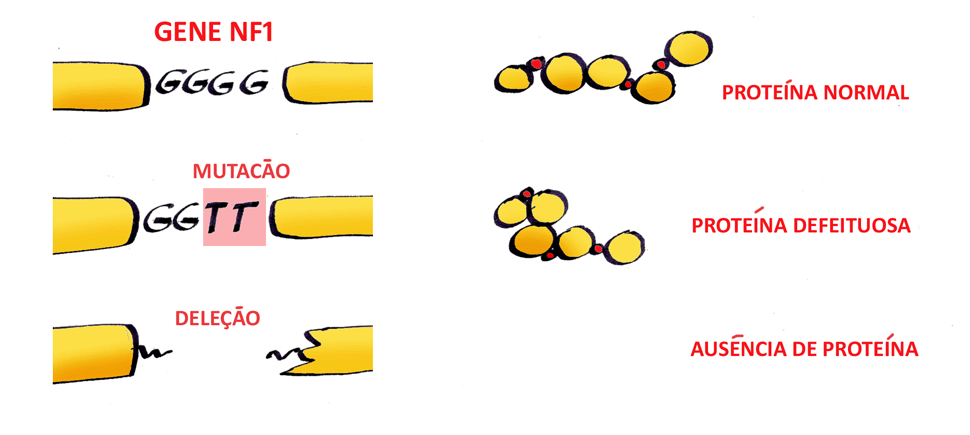
O passo seguinte foi analisarmos a presença da deleção completa do gene NF1 por meio de um teste laboratorial denominado Multiplex Ligation-dependent Probe Amplification (MLPA), o qual foi realizado em 28 pessoas com NF1, entre as quais encontramos 8 delas com a deleção completa e 7 delas apresentam o sinal do segundo dedo dos pés. As demais pessoas com NF1 não apresentavam nem a deleção nem o sinal do dedo.
As análises estatísticas destes resultaram mostraram que a relação entre o sinal do dedo e a deleção foi altamente significativa (P=0,00007). Ou seja, ao encontramos uma pessoa com NF1 e o STS devemos pensar imediatamente na possibilidade de ela apresentar também a deleção completa do gene NF1.
Além disso, todas as pessoas com deleção completa do gene NF1 nesta amostra são do sexo feminino e também são uma mutação nova na família. Ainda não sabemos com segurança o significado deste achado, que será estudado a seguir.
Nossa conclusão é de que o STS é um sinal precoce das manifestações clínicas da NF1 e pode ser útil no diagnóstico e acompanhamento das pessoas que o apresentam.
Agradeço a todas as pessoas que contribuíram para que este conhecimento tenha sido construído em nosso CRNF, em especial às famílias que autorizaram as fotos e coletas de sangue para que o estudo pudesse ser realizado. Vocês contribuíram para que a NF1 seja um pouco mais bem conhecida em todo o mundo a partir desta semana.