Ontem comentei sobre o estudo realizado na Universidade de Indiana nos Estados Unidos, usando o imatinibe em diversas pessoas com NF1 e neurofibromas plexiformes (quem desejar ver o artigo completo em inglês basta clicar AQUI – em inglês).
Outros estudos mais recentes sobre o uso de imatinibe em neurofibromas plexiformes inoperáveis em pessoas com NF1 apontam na mesma direção, ou seja, de que devemos continuar a estudar mais ampla e profundamente esta possibilidade terapêutica.
Algumas reflexões podem ser feitas sobre os resultados do Dr. Robertson e colaboradores, que deram origem ao nosso atual projeto de pesquisa.
A primeira delas é que não sabemos a taxa de crescimento dos plexiformes antes do tratamento com o imatinibe no estudo do grupo do Dr. Robertson. É possível que aqueles tumores com maior taxa de crescimento apresentem resposta melhor ao imatinibe do que os outros menos ativos? Será que a utilização inicial da tomografia computadorizada com emissão de pósitrons (PET CT) poderia definir o nível metabólico dos plexiformes e indicar os mais adequados para o uso do imatinibe?
Segundo, os autores relatam que o tamanho mínimo dos neurofibromas plexiformes deveria ser de 10 mm, ou seja, tumores muito pequenos para causar grandes riscos, de um modo geral. Não seria mais indicado o imatinibe para pessoas com plexiformes maiores (e, de preferência mais ativos, como vimos acima) e inoperáveis?
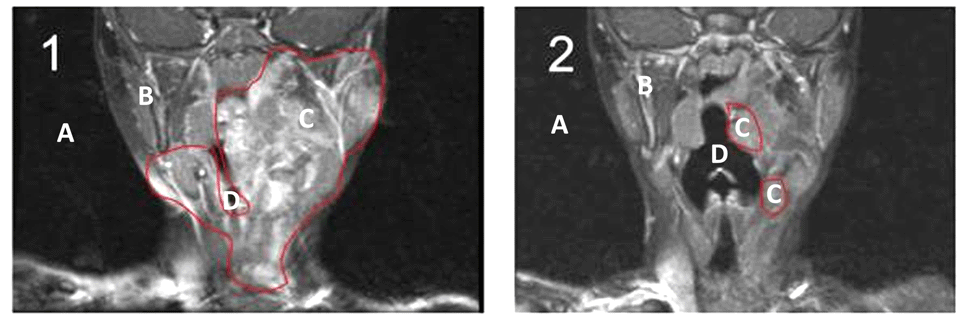
Terceiro, não sabemos se houve uma distinção segura entre os neurofibromas difusos (epineurais) e os neurofibromas nodulares (perineurais), os quais possuem origens embriológicas distintas, diferentes momentos de crescimento, diferentes padrões vasculares e possíveis diferenças nas barreiras teciduais à perfusão do medicamento. Não teria sido prudente a separação prévia ou retrospectiva dos efeitos do imatinibe sobre os neurofibromas difusos e os nodulares?
Quarto, sentimos falta de informação sobre os efeitos do imatinibe sobre os sintomas (como dor e disfunção neurológica) e a qualidade de vida das pessoas com o tratamento experimental. Não teria sido mais útil, para todos nós que trabalhamos na clínica e para as pessoas com NF1, se soubéssemos como o tratamento com o imatinibe foi percebido pelas pessoas que o usaram?
Finalmente, quando se busca corretamente a objetividade (medindo-se apenas o tamanho do tumor) não se corre o risco de perdermos informações que talvez sejam mais importantes para as pessoas com NF1, como dor, outros sintomas e qualidade de vida?
Por exemplo, outra pesquisa, realizada com 3 pessoas com NF1 e plexiformes inoperáveis, mostrou que a intensa dor neuropática (presente em muitos plexiformes, especialmente os nodulares), que é de difícil tratamento, praticamente foi eliminada com o uso de outro medicamento (sirolimus), sem que houvesse redução apreciável do tamanho dos tumores (ver aqui o trabalho completo). Portanto, o tamanho do tumor não é o único problema a ser resolvido.
De qualquer forma, a pesquisa do Dr. Robertson e colaboradores (2012) constitui uma base segura sobre a qual podemos formular a proposta de um novo estudo multicêntrico no Brasil.
Pretendemos realizar um estudo com 50 pessoas com NF1 (25 crianças e 25 adultos), as quais utilizariam o imatinibe em neurofibromas plexiformes inoperáveis e sintomáticos e em crescimento (avaliado pelo PET CT), medindo-se o tamanho do tumor e os efeitos clínicos sobre as pessoas, além de levarmos em conta as diferenças entre plexiformes difusos e plexiformes nodulares.
Estamos dando os passos necessários para este projeto, como submetê-lo aos Comitês de Ética em Pesquisa e buscar o financiamento (cerca de 400 mil reais) junto à Universidade Federal de Minas Gerais, à FAPEMIG e ao CNPq.
Quem sabe, algum leitor deste blog tem recursos financeiros para patrocinar este projeto?