A partir desta quarta-feira, estarei no Congresso Brasileiro de Oncologia, em Foz do Iguaçu, onde farei uma palestra sobre rastreamento de tumores nas neurofibromatoses. Apresentarei o texto abaixo, escrito em colaboração com Nilton, Luíza e Juliana.
Até a próxima segunda.
Rastreamento ou vigilância de tumores nas neurofibromatoses?
Luiz Oswaldo C Rodrigues
Com a colaboração de
Luíza de Oliveira Rodrigues
Juliana Ferreira de Souza
Nilton Alves de Rezende
Centro de Referência em Neurofibromatoses do Hospital das Clínicas
Universidade Federal de Minas Gerais
2015-10-23
Introdução
Aceitei com grande satisfação o convite para realizar esta palestra no Congresso Brasileiro de Oncologia, em Foz do Iguaçu em novembro de 2015, porque para nós, que trabalhamos com as diversas formas de neurofibromatoses, os oncologistas são profissionais fundamentais no manejo clínico deste grupo de doenças genéticas raras.
Com grande probabilidade, apenas entre os brasileiros, metade das 60 mil pessoas com Neurofibromatose do Tipo 1 (NF1) e a maioria das pessoas com Neurofibromatose do Tipo 2 (NF2) e Schwannomatose em algum momento de suas vidas necessitam dos conhecimentos da oncologia para enfrentarem a sua doença.
Para esta palestra, foi proposto o título: “Rastreamento de tumores nas neurofibromatoses”. Como clínico geral, tentarei aplicar às NF aquilo que entendi como o conceito de rastreamento empregado pelos oncologistas e em seguida sugerir algumas especificidades para as doenças com as quais trabalhamos no nosso CRNF.
Rastreamento
Parece-me que o termo rastreamento seriam os esforços realizados na tentativa de identificar tumores suficientemente prevalentes numa determinada população, os quais seriam capazes de causar danos à saúde ou ameaçar a vida caso não sejam precocemente diagnosticados. Além disso, devem ser tumores assintomáticos, mas que, uma vez identificados, possam ser tratados de forma relativamente eficiente, melhorando a qualidade de vida ou a sobrevida das pessoas portadoras do tumor rastreado. Todo este esforço deve apresentar uma relação custo/benefício favorável às pessoas submetidas ao rastreamento, em termos de riscos colaterais e financeiros.
Compreendemos que o termo “rastreamento”, portanto, aplica-se adequadamente a muitos dos tumores que são objeto da atenção dos oncologistas: ou seja, encontrar tais tumores e tratá-los cirurgicamente ou por meio da quimioterapia.
No entanto, nas pessoas com neurofibromatoses precisamos adaptar este conceito de rastreamento, porque: a prevalência de tumores na NF é naturalmente alta; o comportamento natural da maioria deles é benigno; os tumores podem ser sintomáticos ou não; nem sempre há tratamentos disponíveis ou não são necessários para os tumores encontrados; e quando estamos diante de tumores mais agressivos os tratamentos disponíveis não parecem mudar o curso da doença.
Observamos assim que a disposição predominante entre os especialistas em NF diante dos tumores encontrados é mais conservadora, de espera atenta (“watchful waiting”) e principalmente orientada pelos aspectos funcionais e pela qualidade de vida das pessoas com NF.
Isto porque em todas as três formas de NF, além da maioria dos tumores se comportar de forma benigna, sua evolução é imprevisível ao longo da vida. Nas NF, há tumores congênitos e assintomáticos, há aqueles que permanecem décadas sem qualquer manifestação, há aqueles que crescem por algum tempo e se estabilizam por períodos indeterminados, há alguns que regridem espontaneamente sem qualquer tratamento, mas há também uma parte deles que se torna maligna por causas ainda desconhecidas.
Tumores mais comuns nas NF
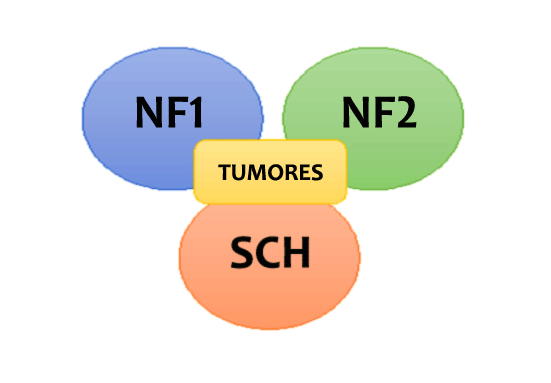
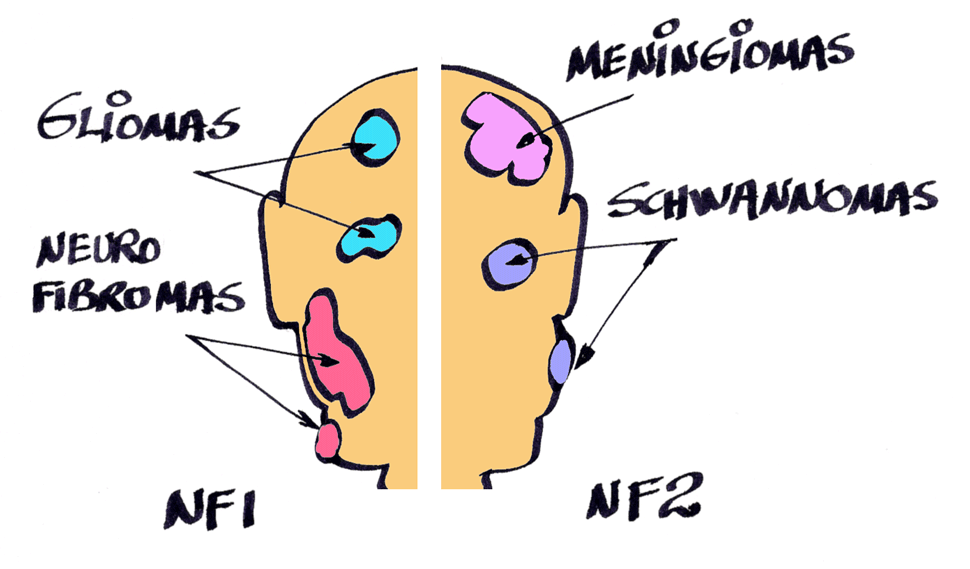
Os principais e mais comuns tumores nas NF são: neurofibromas e gliomas ópticos na Neurofibromatose do tipo 1 (NF1); schwannomas vestibulares e meningiomas na Neurofibromatose do tipo 2 (NF2); e schwannomas dolorosos na Schwannomatose (SCH).
Na NF1, a maioria dos tumores (85% das pessoas os possuem) é formada por neurofibromas cutâneos que crescem lentamente e que precisam ser removidos apenas por razões estéticas, pois jamais se tornam malignos ou ameaçam a vida.
Também na NF1, cerca de 30 a 50% das pessoas têm neurofibromas plexiformes, que são congênitos e histologicamente benignos, mas que podem causar deformidades. Os plexiformes, assim como os cutâneos, podem exigir correção cirúrgica estética ou funcional, e sempre que possível devem ser retirados, mesmo quando benignos.
Uma parte dos plexiformes (10 a 20%) se transforma em tumores malignos da bainha do nervo periférico (TMBNP), que são agressivos e de difícil tratamento. A maior parte dos estudos sobre tratamentos do TMBNP constitui-se de séries de casos, nos quais a sobrevida de 5 anos está em torno de 30%. Assim, a atenção sobre estes tumores concentra-se na identificação de sinais e sintomas precoces que sejam sugestivos de sua possível malignização, como veremos adiante.
Os gliomas ópticos nas pessoas com NF1 são astrocitomas pilocíticos grau I (WHO) e acometem cerca de 15% das pessoas com NF1, mas 85% deles não produzem quaisquer sintomas e requerem apenas acompanhamento clínico. Mesmo os gliomas ópticos que afetam a visão não parecem ser reduzidos pelos tratamentos quimioterápicos atuais nas pessoas com NF1, sendo, portanto, preferível uma atitude mais conservadora e voltada para os aspectos funcionais e não para o tamanho dos tumores. Apenas uma minoria dos gliomas ópticos na NF1 (1,5%) requer abordagens mais agressivas (cirurgia e/ou quimioterapia).
Na NF2, a qualidade de vida e os aspectos funcionais da audição e equilíbrio são os fatores determinantes do momento mais adequado para a tentativa cirúrgica de redução dos schwannomas vestibulares bilaterais (SVB) quando apresentarem aumento na sua velocidade de crescimento. A abordagem cirúrgica deve ser conservadora, buscando-se a redução do tamanho dos SVB muito mais do que a remoção completa do tumor, para se evitar danos colaterais sobre outros nervos cranianos, como o facial e o ramo do próprio oitavo par condutor da audição.
Também na NF2, apenas 40% dos meningiomas apresentam sintomas suficientes para a sua remoção cirúrgica. No momento, ainda não dispomos de tratamentos medicamentosos comprovados tanto para os SVB como para os meningiomas, apesar de alguns estudos em andamento estarem avaliando os efeitos do bevacizumabe sobre os SVB, cujas respostas preliminares ainda não são animadoras. [i]
As pessoas acometidas pela forma mais rara de neurofibromatose, a Schwannomatose, podem apresentar múltiplos schwannomas (exceto vestibulares), dos quais um ou mais pode se tornar doloroso, momento em que está indicada a sua remoção cirúrgica, quando possível. Caso não seja possível sua exérese, existe a opção medicamentosa para o tratamento da dor neuropática.
Princípios fundamentais nas NF
Portanto, podemos compreender que uma regra fundamental nas NF é NÃO SE DEVE RETIRAR OU TRATAR UM TUMOR APENAS PORQUE ELE FOI ENCONTRADO.
Outra postura importante diante de uma pessoa com NF é reconhecermos que apesar de não haver CURA para este grupo de doenças, há diversos TRATAMENTOS E CONDUTAS que melhoram a qualidade e aumentam a expectativa de vida das pessoas acometidas.
Diagnóstico diferencial entre NF1, NF2 e Schwannomatose
A primeira conduta IMPORTANTE é realizar o diagnóstico diferencial entre as três formas: NF1, NF2 e Schwannomatose, para que possamos buscar a presença dos tumores mais comuns em cada uma delas. A identificação adequada do tipo de NF evita que as pessoas a procurem neurofibromas na NF2 ou schwannomas na NF1, o que pode prejudicar os tratamentos.
Realizado o diagnóstico com segurança, o passo seguinte é a identificação dos tumores mais comuns em cada um dos tipos de NF: neurofibromas e gliomas ópticos na NF1, schwannomas vestibulares e meningiomas na NF2 e schwannomas dolorosos na SCH.
Em seguida, devemos avaliar a repercussão clínica e funcional dos tumores. Sugerimos a consulta aos fluxogramas que nos orientam sobre cada um dos principais tumores em cada uma das NF e que estão detalhados e disponíveis em nossa publicação recente no periódico Arquivos de Neuropsiquiatria, de 2015. [1]
Condutas em comum nas NF
A avaliação clínica cuidadosa e anual, acompanhada dos estudos de imagem quando necessários, constituem as ferramentas indispensáveis para o bom acompanhamento dos tumores em todas as formas de NF.
Os estudos de imagem nas NF são preferencialmente realizados com ressonância magnética, mas ocasionalmente a tomografia computadorizada com emissão de pósitrons (PET CT) pode ser necessária na NF1.
É preciso salientar que a PET CT com 18FDG constitui um avanço na diferenciação entre neurofibromas benignos e TMBNP na NF1. Num estudo recente[2], numa série de 42 pessoas com NF1 em nosso ambulatório, a utilização de indicadores quantitativos, semi-quantitativos e qualitativos na avaliação de neurofibromas (geralmente plexiformes ou profundos e mais volumosos) aumentou a sensibilidade para 91%, a especificidade para 90% e o valor preditivo positivo para 98% e o valor preditivo negativo para 69% para a transformação maligna, o que nos tem ajudado imensamente nas condutas terapêuticas.
Tanto na NF1 quanto na NF2 encontramos mais raramente outros tumores, além daqueles expostos acima, que são mais frequentes do que na população em geral, como epiteliomas e astrocitomas encontrados em maior frequência na NF2 e que necessitam de abordagem específica, caso a caso.
Alguns deles necessitam da avaliação de profissionais com experiência no seu tratamento específico e as condutas não diferem daquelas tomadas para indivíduos sem as NF. Por exemplo, 1% das pessoas com NF1 apresentam feocromocitomas, que precisam ser urgentemente tratados dentro dos rígidos protocolos destinados às pessoas sem NF1.
Em pessoas com NF1, o câncer de mama (4 a 6 vezes mais comum nas mulheres com NF1 entre 30 e 50 anos de idade), o tumor sólido gastrointestinal (GIST) em adultos e a leucemia mieloide em crianças são mais prevalentes do que na população em geral, o que nos impõe avaliações regulares para sua detecção precoce.
Outro aspecto a ser considerado na conduta diante dos tumores na NF é o tipo de alteração genética (genótipo) que pode estar relacionado com a gravidade geral do quadro (fenótipo). Por exemplo, numa pessoa com NF1 e com suspeita de deleção do gene, devemos aumentar nossa vigilância sobre os plexiformes, neurofibromas espinhais e profundos por causa de sua maior propensão para a malignização.
Também na NF1, as meninas com glioma óptico parecem evoluir de forma mais grave do que os meninos com NF1 e o mesmo tumor.
No mesmo sentido, na NF2 esperamos mais sintomas e evolução menos favorável dos schwannomas vestibulares e meningiomas quando estamos diante de uma pessoa com a forma sistêmica (herdada ou resultante de mutação em célula germinativa) do que diante de uma pessoa com a forma segmentar (ou em mozaicismo).
Conclusão
As neurofibromatoses exigem de nós, especialistas e oncologistas, a postura de vigilância e acompanhamento diante da evolução imprevisível e um pouco diferente dos seus tumores.
As NF nos fazem reformular aquele conhecido pensamento: que tenhamos recursos para identificar e tratar o que deve ser tratado, que tenhamos capacidade de melhorar e acompanhar o que não deve (ou não pode) ser tratado e que tenhamos sabedoria para distinguir uma coisa da outra.
[2]Hérika Martins Mendes Vasconcelos, 2015. Uso do 18F-FDG PET/CT na suspeita de transformação maligna em indivíduos com Neurofibromatose do tipo 1. Dissertação de Mestrado, Programa de Pós-Graduação do Instituto Nacional de Ciência e Tecnologia de Medicina Nuclear da Universidade Federal de Minas Gerais, Belo Horizonte, MG.
[i] Bevacizumabe na NF2 – A indicação de bevacizumabe vem aumentando e parece-me predominantemente baseada numa revisão feita pelo grupo do Dr. Plotkin, de Boston, Estados Unidos (ver aqui http://www.ncbi.nlm.nih.gov/pubmed/22805104 ).
Em 2012, eles reviram um total de 31 pessoas com NF2 e schwannomas vestibulares que receberam bevacizumabe como opção de tratamento. Vejamos abaixo algumas características das pessoas tratadas, as quais receberam o medicamento durante cerca de 14 meses (6 meses o tratamento mais curto e 41 meses o mais longo).
A idade mediana das pessoas foi de 26 anos, no entanto, havia pessoas de 17 e de 73 anos, o que me deixa um pouco na dúvida se haveria entre elas algumas pessoas com schwannomas vestibulares, mas sem NF2.
A taxa média anual de crescimento dos tumores antes do bevacizumabe era de 64% de aumento, ou seja, um tumor de 2 cm havia passado para um pouco mais de 3 cm em um ano.
Depois de pelo menos 3 meses de tratamento com o bevacizumabe, a melhora na audição aconteceu em 13 de 23 pessoas (57%), ou seja, antes de começar o tratamento a chance do bevacizumabe funcionar seria mais ou menos como jogar uma moeda para cima e escolher cara ou coroa.
Da mesma forma, a redução (20%) do tamanho dos schwannomas na ressonância magnética aconteceu em 17 de 31 pessoas (55%), ou seja, antes do tratamento temos a metade da chance de dar certo.
Mesmo assim, a pequena redução do volume (20%) pareceu mais relacionada com o edema (líquidos ao redor do tumor) do que com a diminuição da parte sólida do schwannoma.
Depois de um ano do tratamento, 90% das pessoas tratadas permanecia com a audição estável. Não entendi bem como compararam com a possibilidade de, se não fossem tratadas, como estaria a audição?
Segundo os autores da pesquisa, o medicamento havia sido “bem tolerado” pelas pessoas.
No entanto, o tratamento com o bevacizumabe não é simples e seus efeitos colaterais podem ser importantes. Por isso, por exemplo, na Inglaterra, duas equipes médicas independentes entre si devem atestar que a pessoa precisa do tratamento com bevacizumabe para que ele seja iniciado.
O bevacizumabe deve ser administrado às pessoas por infusão venosa a cada 15 dias em ambiente hospitalar, o procedimento dura algumas horas e não pode ser dado a pessoas um mês antes ou depois de uma cirurgia ou durante a gravidez e amamentação.
A ressonância magnética do cérebro deve ser repetida a cada 3 meses para controle.
Dias ou semanas depois de iniciado o tratamento podem acontecer quaisquer destes sinais e sintomas: náuseas, febre, alergia cutânea, inchação dos lábios e obstrução da garganta, falta de ar, tontura, tosse contínua, dor no peito e em diversas partes do corpo, fadiga geral, perda do apetite, diarreia ou constipação, aumento da pressão arterial, úlceras na boca, dificuldade de cicatrização, sangramento, embolia pulmonar, baixa resistência às infecções, insuficiência cardíaca, problema no funcionamento renal e infertilidade.
A minha conclusão é que, infelizmente, o bevacizumabe ainda não é uma BOA opção de tratamento. Por enquanto, creio que devemos seguir o tratamento padrão (ver o post de ontem) e torcer para que outra alternativa melhor seja descoberta.