REVISÃO DO MÊS 01 – Avanços terapêuticos para os tumores das neurofibromatoses – Parte 2
Continuo hoje a REVISÃO DO MÊS escrita pelos médicos Jaishri Blakeley e Scott Plotkin, da Faculdade de Medicina de Harvard, e foi publicado na revista Neuro-Oncology de fevereiro de 2016 (ver aqui link para o artigo completo em inglês). Ontem vimos os aspectos gerais das NF, suas semelhanças entre si e com algumas outras doenças. Hoje, resumo algumas das afirmações dos autores sobre a NF1.
Neurofibromatose do tipo 1
Os autores reafirmam a incidência da NF1 em 1 em cada 2500 a 3000 pessoas e, apesar de ser rara, é a mais comum das doenças genéticas autossômicas dominantes do sistema nervoso e das doenças monogenéticas (causadas por um só gene), competindo em frequência com a fibrose cística e com a síndrome do X frágil.
Apesar de ser uma doença que afeta predominantemente a pele e o sistema nervoso central e periférico, a NF1 pode atingir qualquer um dos demais órgãos. As “lesões” cutâneas mais comuns são as manchas café com leite (MCL) as efélides (sardas em áreas do corpo que geralmente não recebem luz solar) e os neurofibromas cutâneos.
As MCL estão presentes desde o nascimento, mas podem ficar mais claras com os anos. Os neurofibromas surgem lentamente no final da infância e continuam a crescer em número e tamanho ao longo da vida. Apesar de causarem transtornos estéticos, eles não ameaçam a vida das pessoas com NF1.
Ao contrário, os neurofibromas plexiformes (NFP) geralmente causam impacto na qualidade de vida e cerca de metade das pessoas com NF1 apresentam um ou mais NFP, que podem surgir desde a raiz do nervo, próximo à coluna, até a sua extremidade em qualquer parte do corpo. Eles podem ser nodulares ou difusos e variar muito em tamanho, envolvendo mais de uma região do corpo, mas aparecem preferencialmente no tronco (31%) cabeça e pescoço (31%) e nas extremidades (25%).
Uma nova informação (pelo menos para mim) foi a afirmação dos autores de que os NFP crescem mais rapidamente durante o começo da infância, atingindo aumentos de 20% ao ano.
Diagnóstico da NF1
É importante reafirmar que os critérios para o diagnóstico da NF1 continuam válidos, apesar de algumas discussões entre os especialistas que sugerem que outros sinais sejam incluídos (hiperintensidades na ressonância magnética e sinal do segundo dedo dos pés, entre outros).
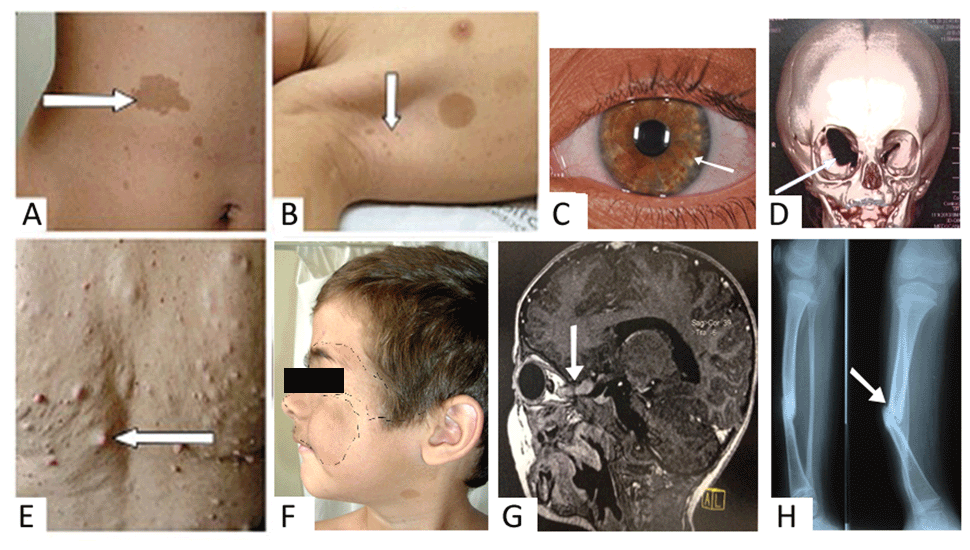
Assim, os sete critérios para o diagnóstico de NF1 continuam sendo (ver figura ao lado):
B) Efélides axilares ou inguinais;
C) 2 ou mais Nódulos de Lisch no exame oftalmológico;
D) Displasia da asa menor do esfenoide;
E) 2 ou mais neurofibromas de qualquer tipo ou
F) 1 neurofibroma plexiforme;
G) Glioma óptico;
H) Displasia de ossos longos, como a tíbia, com ou sem pseudoartrose;
I) Um parente de primeiro grau com NF1 pelos critérios acima.
Tratamentos na NF1
Os autores reafirmam que a cirurgia é a principal base terapêutica para os NFP, lembrando que muitas vezes os cirurgiões não conseguem remover todo o tumor, o que pode resultar e continuidade do crescimento após a cirurgia. Estas dificuldades cirúrgicas e a taxa de crescimento aumentada no começo da infância são as justificativas para as buscas por tratamentos não cirúrgicos dos NFP, como novas substâncias farmacêuticas.
Os NFP podem causar dor, seja ao contato ou espontânea, com características de dor neuropática, e podem levar à disfunção neurológica das áreas afetadas pelo tumor. Além disso, há um risco entre 8 e 13% de um NFP se transformar em tumor maligno, o tumor maligno da bainha do nervo periférico (TMBNP), também denominado de neurofibrossarcoma.
O tratamento dos TMBNP na NF1 deve ser feito imediatamente por resseção cirúrgica completa do tumor com boa margem de segurança, porque, se este objetivo não for alcançado, as opções de tratamento (quimioterapia e radioterapia) oferecem uma sobrevida de 5 anos para menos da metade das pessoas.
Amanhã continuo esta revisão, falando dos gliomas na NF1.
