
Manifesto dos professores de História da Faculdade de Filosofia e Ciências Humanas da UFMG sobre a ocupação estudantil e o quadro nacional.
Nós, professores e professoras do Departamento de História da UFMG abaixo assinados, nos reunimos no dia 1 de novembro de 2016 para discutir a ocupação estudantil na Faculdade de Filosofia e Ciências Humanas e em várias outras unidades da UFMG, bem como para debater outros aspectos da preocupante situação nacional. Por unanimidade decidimos divulgar este manifesto.
Em primeiro lugar, empenhamos total apoio à iniciativa dos estudantes de ocupar prédios da UFMG como forma de protesto contra a Proposta de Emenda Constitucional (PEC) 55, que agora se encontra no Senado, após tramitar na Câmara dos Deputados como PEC 241.
Consideramos que as implicações graves e nefastas da PEC 55 justificam ações de protesto agudas. É preciso chamar a atenção da sociedade para o que está em disputa. O governo alega a intenção de equilibrar o orçamento estatal com um novo regime fiscal, mas a medida provocará, na verdade, a destruição dos serviços públicos, especialmente de educação e saúde.
Desde o início de 2015, os recursos para as universidades públicas já vinham sendo contingenciados, porém, com a ascensão de Michel Temer à Presidência, os cortes foram elevados a uma escala sem precedentes. Problema similar afeta as escolas federais de ensino médio. Não há mais o que cortar, salvo se a intenção fosse a de destruir a educação pública federal de níveis médio e superior com vistas a colocar em prática um projeto de privatização.
Congelando os gastos públicos por 20 anos, sem levar em conta o inevitável incremento populacional e o provável crescimento do PIB, a PEC 55 não trará necessariamente o controle das contas públicas. Certamente, porém, provocará uma redução dramática dos recursos disponíveis para investimentos sociais.
Em lugar de procurar alternativas socialmente mais justas, como reduzir os juros, que oneram a dívida pública e consomem centenas de bilhões de reais do orçamento federal, ou criar impostos, que atinjam também os grandes empresários e as grandes fortunas – e não apenas os assalariados –, a escolha do Governo Federal enfraquecerá os serviços públicos.
Preocupa-nos também o progressivo desmantelamento do sistema de ciência e tecnologia. O atual governo desmontou o Ministério da Ciência, Tecnologia e Inovação (MCTI) e está transformando o Conselho Nacional de Desenvolvimento Científico e Tecnológico(CNPq) em repartição pública secundária, além de impor-lhe uma diminuição drástica de verbas. Já se anunciam cortes profundos no número de bolsas de produtividade e de bolsas de iniciação científica do CNPq para o próximo ano, sem razões de mérito técnico-científico para tanto.
Décadas de investimentos e esforços científicos coletivos podem ser destruídos em poucos meses, tanto mais se a PEC 55 for aprovada. Em relação a essas duas atividades centrais da vida universitária, a educação superior e a pesquisa científica, inúmeras críticas circulam na sociedade, muitas vezes por falta de informações sobre a realidade universitária no Brasil e no mundo. Na comparação com sistemas universitários de outros países, podemos dizer que produzimos muito e fazemos bom uso dos recursos públicos.
O Brasil possui o melhor sistema de pós-graduação da América Latina, que por isso mesmo tem atraído estudantes e professores de vários países, e nossas universidades públicas formam milhares de profissionais todos os anos, com a geração de conhecimento que beneficia o sistema produtivo e dá corpo a programas sociais. As escolas federais de ensino médio, ademais, destacam-se como de excelente qualidade, em todos os mecanismos de avaliação.
Certamente há problemas a enfrentar e o desempenho das universidades e instituições de pesquisa pode ser aprimorado. Porém, não será cancelando os investimentos públicos em educação e ciência, além da saúde, que o Brasil irá encontrar boas saídas. Bem ao contrário, o futuro pertencerá aos países que tiverem capacidade de produzir conhecimento, para gerar riquezas e também para refletir com inteligência sobre o seu uso equilibrado.
Manifestamos preocupação, também, com a Medida Provisória nº 746/2016, que promove alterações no Ensino Médio; com a proposta do Programa Escola Sem partido; e com os episódios de violência que têm acontecido em algumas escolas públicas do ensino básico, igualmente ocupadas por estudantes, em que autoridades policiais têm abusado da força e desrespeitado direitos individuais, tendo o mesmo efeito a atuação de determinados movimentos que, frequentemente, agem como milícias.
Estaremos atentos para que isso não ocorra nos espaços da UFMG, convictos de que as disputas políticas devem ser travadas nos limites do respeito às diferenças e às garantias legais, características inerentes às verdadeiras democracias.
Professores presentes à reunião de 01 de novembro de 2016, signatários da nota:
Adriana Romeiro
Adriane Aparecida Vidal Costa
Alexandre Almeida Marcussi
Ana Carolina Vimieiro Gomes
Ana Paula Sampaio Caldeira
André Luis Pereira Miatello
Betânia Gonçalves Figueiredo
Douglas Attila Marcelino
Ely Bergo de Carvalho
Heloísa Maria Murgel Starling
José Antônio Dabdab Trabulsi
José Newton Coelho Meneses
Luiz Carlos Villalta
Luiz Duarte Haele Arnaut
Magno Moraes Melo
Mariana Silveira Moraes
Miriam Hermeto de Sá Motta
Priscila Carlos Brandão
Regina Helena Alves da Silva
Regina Horta Duarte
Rodrigo Patto Sá Motta
Professores que se tornaram signatários da nota após a reunião:
Adalgisa Arantes Campos
Cristina Isabel Campolina de Sá
Eduardo França Paiva
João Pinto Furtado
Kátia Gerab Baggio
Mauro Lúcio Leitão Condé
Rafael Scopacasa
Tarcísio Rodrigues Botelho
Vanicléia Silva Santos
Este blog tem divulgado informações científicas sobre as neurofibromatoses em linguagem compreensível para milhares de famílias que o acessam em busca de apoio para o enfrentamento cotidiano destas doenças genéticas raras.
Somente é possível oferecer estas informações porque sou médico formado pela Faculdade de Medicina da Universidade Federal de Minas Gerais, onde sou professor há mais de 40 anos. Além disso, o Hospital das Clínicas desta mesma Universidade acolheu nosso Centro de Referência em Neurofibromatoses, onde atendemos milhares de pessoas de todo o Brasil pelo Sistema Único de Saúde.
Esta mesma Universidade e o Sistema Único de Saúde agora se encontram ameaçados pela PEC 241 do Governo Temer, que limita de forma absurda os investimentos sociais por 20 ANOS!
Se a Universidade Pública Brasileira e o Sistema Único de Saúde forem sucateados em benefício das empresas privadas de ensino e de medicina, nosso Centro de Referência em Neurofibromatoses desaparecerá com ela.
Portanto, em defesa da Universidade, do SUS e do Centro de Referência em Neurofibromatoses me uno às pessoas que estão protestando contra a PEC 241.
Em solidariedade aos milhares de estudantes e professores que estão ocupando as escolas e universidades pelo Brasil afora, a partir de hoje este blog oferecerá apenas informações sobre os protestos contra a PEC 241.
Belo Horizonte, 2 de novembro de 2016
Dr. Lor

Representei a Associação Mineira de Apoio às Pessoas com Neurofibromatoses (AMANF) no II Congresso Ibero-Americano de Doenças Raras em Brasília, onde realizei uma palestra sobre a rede humana necessária para o cuidado das pessoas com neurofibromatoses (ver aqui o resumo da palestra AQUI).
Primeiramente, quero parabenizar a equipe da Associação Maria Vitória de Doenças Raras (AMAVI) sob a liderança da sua presidente Lauda Santos, pelo excelente trabalho de programação, pela dedicação à causa e pela hospitalidade.
Acompanhei muitas das palestras e mesas redondas, que me permitiram aprendizados diversos sobre vários assuntos. Desde informações sobre as políticas públicas do Ministério da Saúde até os projetos empresariais para as doenças raras, além de informações sobre doenças raras que eu desconhecia completamente.
Entre minhas impressões, notei que uma das frases mais repetidas em todos os eventos sobre doenças raras é aquela que diz que, segundo as Organização Mundial de Saúde, já teriam sido identificadas cerca de 5 a 8 mil doenças raras e que cerca de 3 a 6% da população mundial seria afetada por doenças raras.
Parece-me que precisamos melhorar a precisão estatística desta informação, porque entre 5 e 8 mil doenças existe uma margem de erro de 60% e entre 3 e 6% de pessoas afetadas existe uma variação de 100%. Ou seja, esta imprecisão nos dados sobre as doenças raras enfraquece os argumentos dos defensores das pessoas afetadas, por exemplo, numa discussão de políticas públicas destinadas a elas. Por outro lado, temos que procurar saber se estes números têm sido inflacionados por interesses comerciais.
Assim, uma de nossas tarefas importantes para o futuro é dimensionarmos de forma realista o verdadeiro número de doenças raras e sua incidência na população.
Outra impressão que tive no Congresso foi a dominância dos temas discutidos em função dos medicamentos para algumas doenças raras. As palestras mais concorridas e com maior carga emocional envolvida foram aquelas que abordaram o fornecimento de alguns medicamentos pelo Sistema Único de Saúde (SUS), a liberação de medicamentos pela Agência Nacional de Vigilância Sanitária (AVISA) e os processos judiciais que as famílias têm realizado para obter determinados medicamentos (judicialização).
No entanto, sabemos que a grande maioria das doenças raras não possui ainda qualquer medicamento cientificamente comprovado e que deva ser ministrado às pessoas afetadas.
Esta imensa maioria de doenças raras precisa de outros recursos, especialmente a formação de equipes multidisciplinares que sejam capazes de fornecer o diagnóstico correto, aconselhamento genético, fisioterapia, fonoaudiologia, terapia ocupacional, enfermagem, enfermagem, apoio psicológico e psicopedagógico.
Ou seja, a maioria das doenças raras necessita de uma tribo inteira de cuidadores para que a qualidade de vida das pessoas afetadas seja melhorada, mesmo que não haja cura para a doença.
No entanto, a discussão sobre medicamentos abafa a criação desta rede pública de suporte às pessoas com doenças raras. Uma das consequências desta prioridade seletiva sobre medicamentos é que a estrutura multidisciplinar dos Centros de Referência em Doenças Raras que deveriam existir a partir da Portaria 199 do Ministério da Saúde de 2014 praticamente não saíram do papel.
E por que apenas algumas doenças raras recebem mais atenção do que a maioria das outras? Porque algumas delas já possuem medicamentos fabricados por laboratórios farmacêuticos que nos percebem apenas como um tipo de mercado de consumidores.
Esta postura empresarial em busca do lucro ficou explícita numa das palestras, a realizada pelo Dr. Fernando Ferrer da Multinational Partnerships, que terminou sua fala convidando os empresários multinacionais da indústria farmacêutica a investirem na América Latina, pois aqui seria uma excelente oportunidade de negócios no campo das doenças raras.
Creio que estas divergências nas doenças raras se encaixam na luta política entre o modelo de saúde pública (universal, para todos, e portanto estatal) e o outro de saúde privada (apenas para os que podem pagar).
Esta é a grande luta política sobre a saúde que ocorre neste momento em todo o mundo e, em particular, no Brasil, onde o governo Temer quer limitar os investimentos públicos em benefício da sociedade para maior lucro dos bancos e empresas multinacionais.

Na próxima postagem comentarei o Congresso de Doenças Raras que aconteceu neste último final de semana em Brasília.
Hoje retomo uma questão muito importante: a medida da pressão arterial nas pessoas com NF1.
Tenho comentado a importância da medida periódica (no mínimo anual) da pressão arterial em todas as pessoas com NF1, que deve ser realizada desde a infância (com aparelhos de medida adequados ao seu tamanho) até a vida adulta (ver aqui: AQUI ).
Isto porque, como já foi comentado, sabíamos que as pessoas com NF1 correm maior risco de apresentarem dois problemas que causam aumento da pressão arterial: displasia da artéria renal (com estenose ou obstrução do fluxo de sangue para um ou os dois rins) e feocromocitoma (um tumor capaz de produzir adrenalina).
Portanto, devemos ficar atentos à pressão arterial de pessoas com NF1 porque estes dois problemas são potencialmente fatais e podem ser curados com tratamentos adequados.
No entanto, ainda não sabíamos se, em geral, as crianças com NF1 apresentam pressão arterial mais aumentada do que a população infantil sem a doença. Foi justamente esta pergunta que um grupo de pesquisadores de Israel tentou responder com seu estudo, cujos resultados foram recentemente publicados (ver aqui artigo em inglês: AQUI ).
O grupo orientado por Shay Ben-Shachar mediu a pressão arterial 3 vezes em 224 crianças com NF1, metade meninas, com idade em torno de 9 anos. Os resultados da pressão arterial foram classificados de acordo com a idade, o sexo e o percentil da altura de cada uma delas, em normais (menor do que o percentil 85%), pré-hipertensos (entre os percentis 85 a 95%) e hipertensos (maior que o percentil 95%).
Os resultados mostraram que em torno de 13% das crianças com NF1 eram pré-hipertensas e outras 13% eram hipertensas, o que significa que a hipertensão arterial é dez vezes mais frequente em crianças com NF1 do que na população em geral.
Outro achado importante do estudo foi que a hipertensão foi ainda maior na primeira medida (20%) do que na terceira, por isso devemos repetir a medida quando encontramos valores altos da primeira vez.
Os autores observaram mais alterações no sistema urinário avaliado pelo ultrassom, mas as condições do estudo não permitiram aos autores confirmar ou excluir as alterações vasculares nas crianças com hipertensão.
Assim, ainda não sabemos a definição exata da causa da hipertensão arterial nas crianças com NF1, mas eles levantaram a hipótese de que a pressão arterial aumentada faça parte das características da NF1. Esta hipótese merece ser mais estudada no futuro.
De qualquer forma, este estudo confirma e aumenta a necessidade de vigilância sobre a pressão arterial de pessoas com NF1, pois se não for tratada a hipertensão arterial pode causas complicações, inclusive fatais.
Portanto, não se esqueça de medir sua pressão arterial pelo menos uma vez por ano.

II Congresso Ibero-americano de Doenças Raras
Acontecerá em Brasília no próximo final de semana (21 e 22 de outubro de 2016) o II Congresso Ibero-americano de Doenças Raras, no Centro de Convenções Ulysses Guimarães.
Ver a programação completa no endereço: www.amaviraras.com
Aceitamos o convite para falar sobre as neurofibromatoses e assim apresento abaixo o resumo da palestra que farei no sábado às 16 horas.

As neurofibromatoses (NF) são doenças genéticas que atingem cerca de oitenta mil pessoas no Brasil e que fazem parte do grande grupo de doenças raras.
As NF constituem três doenças distintas, NF1, NF2 e Schwannomatose que possuem com comum o aparecimento de tumores no sistema nervoso e na pele. No restante de sua apresentação, cada uma das NF é completamente distinta das demais.
A mais encontrada delas, a NF1, geralmente apresenta-se desde a infância e sua manifestação mais comum são as dificuldades de desenvolvimento psicomotor. A NF2, mais rara, geralmente é diagnosticada na juventude e acomete a audição e o equilíbrio. A Schwannomatose é mais rara ainda e mais tardia, e seu principal sintoma é a dor.
A medicina ainda não tem cura para as NF, mas todas elas possuem tratamentos que podem melhorar a qualidade e a duração da vida das pessoas afetadas.
A principal demanda das pessoas e famílias afetadas pelas NF é por informação confiável, porque as doenças raras, apesar de afetarem cerca de 3% da população mundial, não podem ser compreendidas em profundidade por todos os profissionais da saúde.
Diante disso, nossa entidade de apoio às pessoas com NF, a Associação Mineira de Apoio às Pessoas com Neurofibromatoses (AMANF), tem procurado suprir esta demanda com as seguintes ações:
1) Informação pessoal durante os atendimentos clínicos no Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais (cerca de 1 mil famílias cadastradas em 12 anos de existência);
2) Reuniões mensais de pessoas com NF, suas famílias e profissionais de saúde (que vêm ocorrendo há 14 anos de forma ininterrupta);
3) Publicação e distribuição de cartilhas didáticas, escritas em linguagem acessível (por exemplo, a cartilha “As manchinhas da Mariana” está na terceira edição, com tradução para o inglês, espanhol e alemão e cerca de 5 mil exemplares distribuídos);
4) Palestras educativas em congressos da área de saúde (como este II Congresso Ibero-Americano de Doenças Raras), em escolas e instituições públicas;
5) Publicação de informações em página na internet;
6) Manutenção de blog na internet com sistema de perguntas e respostas (cerca de 300 temas já abordados, com média de 12 mil acessos mensais);
7) Apoio a pesquisas de alunos de iniciação científica (5), mestrado (8) e doutorado (4) nas áreas de medicina, fonoaudiologia, psicologia, nutrição, biologia molecular e fisioterapia;
8) Publicação de artigos científicos (18) e participação em congressos nacionais (8) e internacionais (32);
9) Curso de capacitação de profissionais e familiares no cuidado de pessoas com neurofibromatoses.
10) Movimentos junto a entidades políticas para promoção do reconhecimento legal das necessidades especiais das pessoas afetadas pelas NF.
Além disso, uma parte das pessoas com NF apresenta dificuldades de inserção social, o que faz com que elas necessitem de apoio de familiares e amigos para levarem uma vida adequada.
Esta rede de apoio precisa ter origem na pessoa afetada pela NF, em função de sua maior ou menor necessidade de cuidados especiais.
Em seguida, é claro, os familiares formam o primeiro elo da pessoa com NF e a sociedade, seguindo-se os amigos, que devem ser conscientizados dos problemas e dificuldades das pessoas afetadas pela NF, para que possam compreender melhor seu comportamento.
Os professores e a escola constituem o passo seguinte, que devem receber laudos e cartilhas que possam auxiliar os profissionais da educação a exercerem o seu papel de desenvolvimento do potencial humano das pessoas com NF, especialmente a NF1.
O nível seguinte de apoio deve ser formado pelos profissionais da saúde no bairro da pessoa afetada pela NF, os quais precisam ser informados da existência e característica da doença por meio das cartilhas especializadas e dos endereços adequados na internet. O Programa de Saúde da Família, os Postos de Atendimento e os demais segmentos do Sistema Único de Saúde devem ser mobilizados progressivamente para o atendimento das pessoas com NF.
Esta rede humana de apoio às pessoas com NF pode ser fortalecida pela participação em associações como a AMAVI, a AMANF e outras, que precisam abraçar o seu papel político na formação de uma representação social da doença junto à sociedade e, desta forma, influenciar as decisões coletivas nas políticas públicas de bem-estar social e direitos humanos.
A imagem social construída para as NF poderá influenciar a política nacional de saúde, a política de formação de recursos humanos destinados ao seu cuidado, assim como o financiamento para pesquisas científicas que venham a contribuir para mais qualidade de vida para as pessoas afetadas.
O círculo virtuoso desta rede humana de apoio às pessoas com NF é formado por autoestima, amor familiar, amizade, empatia profissional, espírito público, justiça social e solidariedade.
Brasília, outubro de 2016.
Dr. Luiz Oswaldo Carneiro Rodrigues
Professor Titular
Coordenador Clínico do Centro de Referência em Neurofibromatoses
Hospital das Clínicas da Universidade Federal de Minas Gerais
Caras leitoras e leitores,
Atingimos mais de 300 assuntos relacionados com as neurofibromatoses já tratados neste blog diariamente.
A maioria das perguntas que tenho recebido ultimamente já foram respondidas e podem ser recuperadas escrevendo-se o assunto na caixa de perguntas à direita desta página.
Outras perguntas são referentes a problemas pessoais ou muito especiais para serem tratados publicamente no blog. Como sabem, somente podemos opinar diretamente sobre a saúde de uma pessoa depois de a examinarmos clinicamente.
Diante disso, decidi mudar a publicação de diária para semanal, a não ser que surjam informações que mereçam ser divulgadas imediatamente.
Vamos experimentar assim por dois meses e veremos como funciona.
Retornarei em 17 de outubro.
Até lá.
Dr. Lor

“Vim só com minha mãe das outras vezes, porque ela que sempre fica do meu lado, nos exames segura minha mão, falo: Tá bem mãe, ela faz um carinho, solta, foi ela que descobriu as coisas na internet, chorou muito depois alegrou um pouco, porque agora a gente sabe o que você tem, ela falou enxugando os olhos, voltamos hoje no mesmo doutor, desta vez meu pai veio, primeira vez que vem, botou até sua camisa de manga comprida, fiquei mais nervoso, ele queria conferir, saber a verdade, confirmar, porque sempre achou que eu não fazia o que ele mandava porque era preguiça, porque eu não queria, ele não via a dificuldade, Se você esforçar, consegue, olha seu irmão, mais novo que você!, meu irmão não tem problema, tá na escola e a turma vive lá em casa chamando ele para o futebol, eu nunca chamam, nem aprendi a andar de bicicleta, Você não sabe o que é se esforçar, se esforçar, se esforçar e não conseguir nunca, pensei em dizer, mas tive medo dele zangar mais, meu pai parece com vergonha de mim na frente dos outros, Fala direito, arruma essa língua, menino!, brigou com o Demerval porque Demerval contou uma piada de fanho na frete dele, você está gozando meu filho, Demerval?, voltamos para casa e meu pai me botou lendo em voz alta uma revista, de novo, fala claro, levanta a voz, vamos, você consegue, se esforça, anda!, depois de um tempo jogou a revista longe e se trancou no quarto, nunca mais falou com o Demerval, depois quando não entendia o que eu falava mandava repetir até ele escutar direito, fala todas as letras pela boca e não pelo nariz!, Mas é pro bem dele que estou cobrando, se defendia quando minha mãe reclamava que eu já estava chorando e ele não me dava sossego, rasgava meus cadernos com a letra que ele não entendia, faz de novo, gritava, enquanto não ficar bom você vai repetir, como é que você quer ter um emprego na vida com esses garranchos?, eu ia só piorando a letra com o medo de tomar um tapa e a tinta borrava com o choro, não adianta chorar e escreve de novo, vamos, se esforça, minha mãe vinha me socorrer, já falei que precisamos levar o menino na psicóloga, a professora já falou, todo mundo, menos você que não entende que seu filho precisa de ajuda, Você é que protege ele e assim ele nunca será um homem de verdade, será sempre esse banana, chorei mais ainda dele me chamar de banana, aí minha mãe conseguiu marcar nas clínicas e fomos nesse doutor que disse: é da doença, dos problemas de nascimento, precisa mesmo de ajuda, minha mãe voltou para casa com os pedidos de exame e meu pai não disse nada pela primeira vez, as dores foram aumentando, na segunda vez tive que fazer outro exame porque parecia câncer mas não era, aumentou o remédio de dor, parei de sair de casa, ficava dormindo quase o dia todo, mexer para qualquer coisa doendo mais do que antes, marcou para operar mas não tinha jeito, parece que tem um monte de caroço por dentro e por fora, não tem cura, vamos tentar parar de doer com remédio, disse o doutor que ia operar, voltamos com o tal relatório, carta para a escola, mas hoje meu pai quis vir pessoalmente, escutou tudo, perguntou umas coisas, mas não compreendi direito tudo que disseram, minha mãe ficou com os olhos cheios d’água, mas entendi que é para aproveitar a vida que eu tenho hoje, deixar eu brincar com o celular e comer o que eu mais gostar, meu pai foi ficando diferente, é da doença, então, ele disse e olhou para mim diferente do seu jeito de sempre me olhar, como se estivesse me vendo pela primeira vez na vida, até espantei quando ele levantou da cadeira e me abraçou, desculpe, meu filho, eu não sabia, meu coração está batendo que dói na cabeça e não sei o que falar, mas nunca mais vou esquecer este abraço”.
Acho que foi isso que entendi no olhar daquele rapaz abraçado com seu pai, na sala do nosso ambulatório do Centro de Referência em Neurofibromatoses.
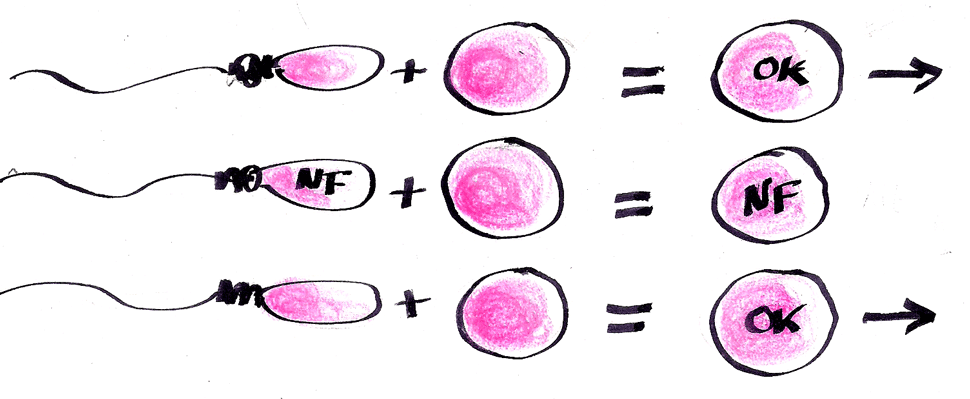
“Sou portadora da NF1. Tenho 35 anos e não tenho filhos, gostaria de saber quais são os reais riscos de uma gravidez considerando essas condições, ou se ainda existe uma possibilidade de uma fertilização in vitro usando óvulos sadios.” EJFP, da Bahia.
Cara E. Obrigado pela sua pergunta.
Os riscos mais comuns para a gravidez em mulheres que apresentam a NF1 já foram discutidos neste blog (ver AQUI).
Quanto à fertilização artificial (in vitro), sim, já existe a técnica para a geração de crianças sadias de um dos pais com NF1 e defendemos que esta técnica seja implantada pelo SUS nos hospitais de Referência . Enquanto isto não acontece, podemos compreender melhor a legislação brasileira sobre este assunto.
No Brasil, a reprodução assistida (RA) é fiscalizada pelo Código de Ética Médica do Conselho Federal de Medicina e pela Lei Federal de Biossegurança (Lei 11.105/05). Todas estas normas orientam médicos e pacientes sobre a RA e as questões envolvidas, como número de embriões transferidos em cada tentativa, doações de óvulos e espermatozoides, barriga de aluguel, conservação em congelamento dos embriões e outras questões envolvendo material genético.
Estas leis pretendem garantir a todos os brasileiros o direito à RA e torna obrigatório o esclarecimento dos futuros pais por parte dos profissionais da saúde, de forma clara, objetiva e o recolhimento da assinatura dos interessados, inclusive dos doadores de material genético, depois de todas as pessoas se sentirem bem informadas.
Algumas normas gerais devem ser observadas: a) deve haver algum problema para a gestação natural (não assistida); b) deve ser conhecida a probabilidade de sucesso na técnica de RA que se pretende empregar; c) não pode haver risco grave para a saúde da pessoa ou do possível descendente; e d) a idade máxima para a RA é de 50 anos para mulheres se engravidarem e de 35 anos para doação de óvulos e de 50 anos para a doação de espermatozoides.
Algumas restrições à RA existem: a) não pode ser usada para a escolha do sexo do bebê ou qualquer outra característica biológica, a não ser para se evitar alguma doença conhecida nem um dos pais (como no caso das neurofibromatoses); b) não se pode fecundar óvulos humanos a não ser para a procriação; c) não pode haver lucro nem comércio na doação de material genético (espermatozoides ou óvulos); d) os doadores não devem conhecer os futuros pais e nem ao contrário; e) e diversas outras normas orientadoras que podem ser acessadas na lei acima referida.
Comentei acima apenas alguns dos aspectos envolvidos na legislação brasileira sobre RA para lembrar que esta questão do controle ético de como a humanidade irá gerar a sua continuidade é um dos temas mais importantes em discussão.
Os cuidados que devemos ter com a manipulação genética de seres humanos são necessários para evitarmos que esta técnica de reprodução assistida seja usada para outras finalidades que podem comprometer o futuro da humanidade.