Comentei recentemente que a chance de um casal sem NF1 ter um filho com NF1 (mutação nova) é de 1 em 6 mil, mas esta informação é um pouco diferente daquela que venho repassando às famílias e que estão em diversas publicações, inclusive neste blog.
Somente me dei conta deste engano ao refazer as contas para calcular a chance de um casal vir a ter gêmeos com NF1. Vamos rever juntos o cálculo correto para um casal sem NF1 ter um filho ou filha com NF1.
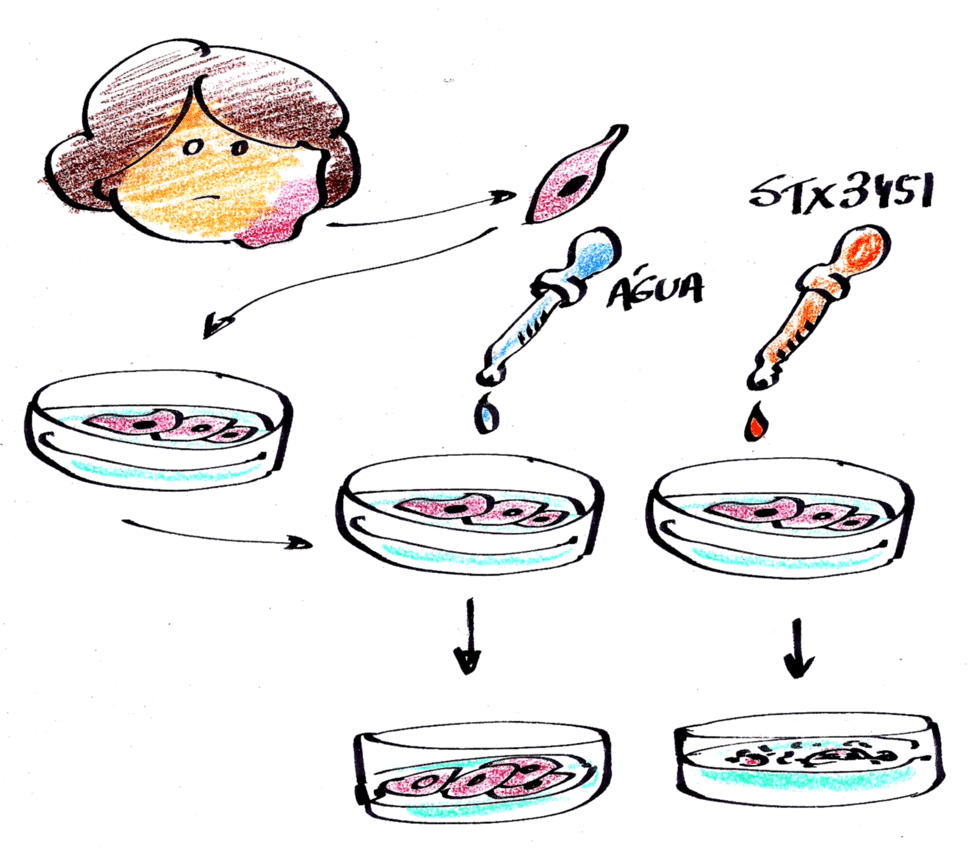
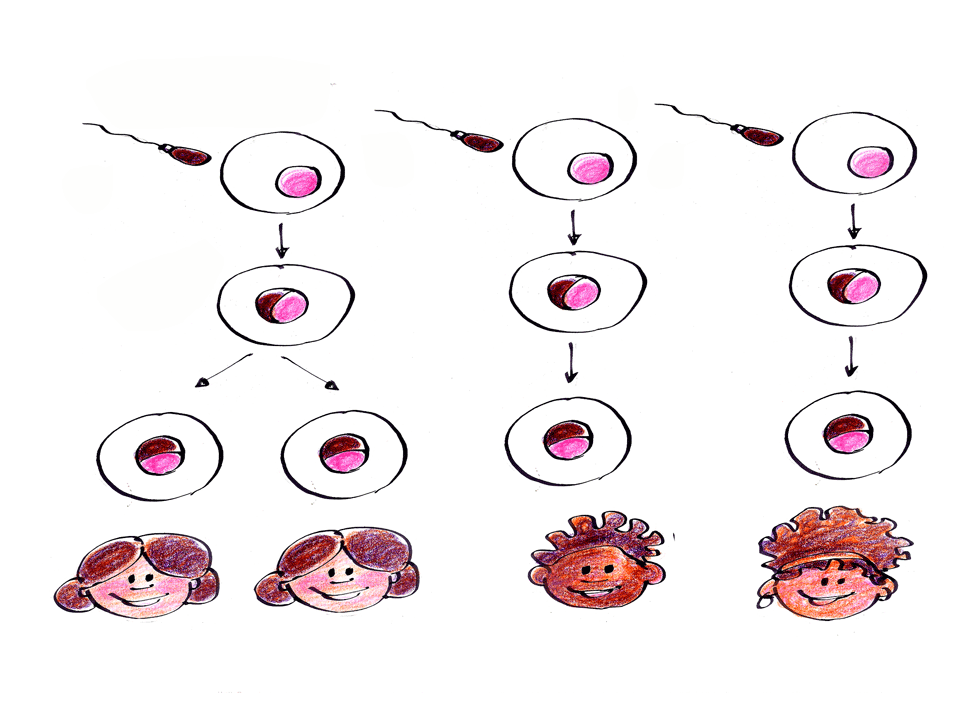
A incidência de NF1 em qualquer população é de aproximadamente 1 em cada 3 mil crianças nascidas. Sabemos que metade das crianças com NF1 herdou a mutação no gene de um dos seus pais que já possui a doença. A outra metade das crianças com NF1 tem a doença por causa de mutações novas, ou seja, são variações genéticas que acontecem por acaso, no momento da formação do espermatozoide ou do óvulo, e que podem causar erros na produção de uma proteína chamada neurofibromina, a qual é responsável pelo controle do crescimento das células e pelo desenvolvimento do bebê (ver a ilustração na cartilha “As manchinhas da Mariana” clique AQUI).
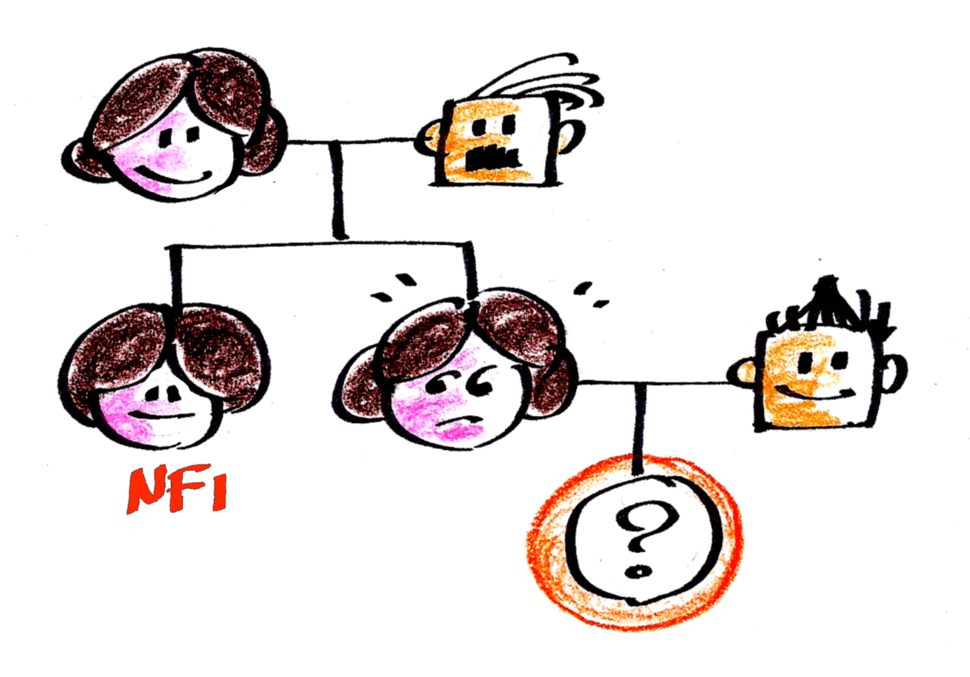
Assim, naquelas 3 mil pessoas da população que precisamos reunir para encontrar uma pessoa com NF1, apenas “meia” pessoa seria resultante de uma mutação nova para a NF1. Como não existe “meia” pessoa, precisamos de reunir o dobro de pessoas na população para encontrarmos uma “pessoa inteira” com mutação nova, ou seja, precisamos reunir 6 mil pessoas na população.
Assim, a chance de um casal sem NF1 vir a ter um filho (ou filha) com NF1 (mutação nova) é de 1 para 6000. Por outro lado, a chance de um casal em que um dos pais possui NF1 vir a ter um filho (ou filha) também com NF1 (mutação herdada) é de 1 para 2, ou seja 50%.
Este cálculo mostra uma probabilidade um pouco maior de mutação nova em pessoas que não tem NF1 (1 para 6 mil) do que as pesquisas citadas pelo Dr. Friedman no livro do Riccardi (1999), que encontraram 1 mutação nova em cada 7800 a 23000 gametas, ou seja, espermatozoides ou óvulos (esta variação é por conta de diferenças de métodos entre os estudos).
Uma possível explicação para meu cálculo ter resultado numa maior chance (1 para 6 mil é maior do que 1 para 7800) talvez esteja no fato de que as pessoas com NF1 têm menor número de filhos do que a população em geral.
Considerando que pais com NF1 (especialmente os homens com NF1 que têm menos de 50% de filhos em relação aos homens sem NF1), isto aumentaria a participação das mutações novas no conjunto de pessoas que encontramos com NF1. Em outras palavras, por isso encontramos metade das crianças com NF1 com mutação nova e a outra metade com mutações herdadas.