
Dando prosseguimento à nossa divulgação online dos capítulos escritos para a Edição Comemorativa dos 20 anos do CRNF, a ser lançada em dezembro de 2024, apresentamos o texto fundamental da Dra. Luciana Baptista Pereira, professora de Dermatologia na Faculdade de Medicina da UFMG, com informações muito importantes que faltavam em nosso site, o que vem ampliar nosso conhecimento sobre a NF1. Muito obrigado, em nome de toda a equipe do CRNF.
Dr. Lor
Professora Luciana Baptista Pereira
Dermatologista e Professora da
Faculdade de Medicina da UFMG
MANCHAS CAFÉ COM LEITE
As manchas café com leite (MCL) estão presentes no primeiro ano de vida em 99% das crianças que posteriormente serão diagnosticadas com neurofibromatose do tipo 1 (NF1). São o sinal mais comum e mais precoce, seguidas pelas efélides axilares ou inguinais (Nunley et al. 2009). Apesar disso, as MCL não são patognomônicas da NF1. Uma única MCL está presente em cerca de 2,5% dos neonatos em estudos populacionais (Shah 2010) e, em um estudo brasileiro, percentual semelhante foi encontrado: as MCL foram encontradas em 2,8% dos recém-nascidos examinados nas primeiras 36 horas de vida. (Pereira 1999)
As MCL podem aumentar em número até os 2 a 4 anos de idade, sendo incomum surgirem após os 6 anos a não ser em pacientes com NF1, cujo número pode aumentar até a adolescência e vida adulta. (Boyd 2010; Shah 2010) A possibilidade das MCL estarem presentes de forma isolada, sem associação com outras alterações, é inversamente proporcional ao seu número. Uma criança com 3 ou mais MCL deve ser avaliada cuidadosamente pela possibilidade de haver associação com síndromes, pois a presença de mais que 3 MCL são detectadas em apenas 0,3% das crianças sem evidência de desordem genética. (Nunley et al. 2009) Quanto maior for o número de MCL, maior a probabilidade do diagnóstico ser NF1. A presença de seis ou mais MCL maiores que 5 mm em indivíduos pré-puberais e 15 mm em adultos é um dos critérios diagnósticos da NF1.
Além do número, as características morfológicas das MCL são importantes para o diagnóstico. As MCL se tornam visíveis ao nascimento ou nas primeiras semanas de vida. A cor geralmente é uniforme, variando do marrom claro ao escuro. A cor pode variar entre as manchas do mesmo indivíduo e entre indivíduos diferentes, tendendo a ser mais escuras nos negros e mais claras nas pessoas brancas. (Shah 2010) (Friedman 2016)
As MCL são maculares, sem nenhuma alteração do relevo. Se houver alguma alteração no relevo, o diagnóstico de neurofibroma plexiforme se torna mais provável. (Friedman 2016) – Figura 1 A – As máculas hipercrômicas com alteração de relevo subjacente no tórax em paciente com NF1 – o diagnóstico é neurofibroma plexiforme e não, mancha café com leite. Manchas café com leite podem ser vistas no abdome
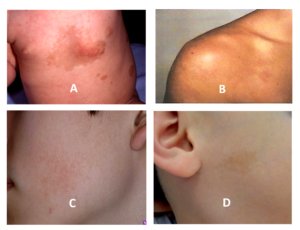
Figura 1 – Lesões hipercrômicas. A – Neurofibroma plexiforme; B e C: Nevos de Becker mácula hipercrômica com bordas irregulares encimadas por micropápulas: a alteração de relevo afasta o diagnóstico de mancha café com leite; D: Mácula hipercrômica também chamada de mancha café com leite atípica.
O tamanho geralmente varia de 5 a 30 mm de diâmetro, mas podem ser maiores chegando a 20 cm. Crescem de forma proporcional ao crescimento da criança. A distribuição parece ser randomizada, poupando apenas o couro cabeludo, palmas e plantas, e são menos comuns na face. A cor e a distribuição das MCL não apresentam relação com a exposição solar (Boyd et al., 2010).
As MCL típicas da NF1 são ovaladas e apresentam bordas regulares, mesmo quando de tamanho maior. As manchas hipercrômicas que apresentam bordas irregulares, denteadas, são denominadas de MCL atípicas por não serem características da NF1. (Nunley 2009; Kehrer-Sawatzki 2022) Estas máculas hipercrômicas de bordas irregulares, denteadas, chamadas de MCL atípicas por vários autores, (Figura 1 D) são também denominadas de lesões hipermelanóticas segmentares e refletem um mosaicismo pigmentar e quando pequenas, são denominadas de nevos hipercrômicos. (Torrelo 2005) Essas lesões hipercrômicas de bordas irregulares correspondem ao inverso do nevo despigmentado.
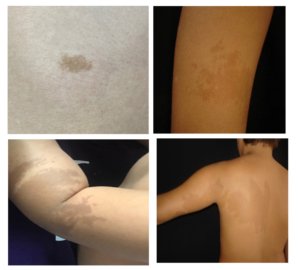
Figura 2 – Outras lesões hipercrômicas: nevos hipercrômicos – também denominados de manchas café com leite atípicas – são máculas hipercrômicas de bordas irregulares, denteadas, segmentares – são o inverso do nevo despigmentado, também denominadas de lesões hipermelanóticas segmentares.
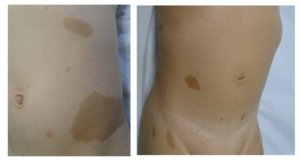
Figura 3 – Manchas café com leite típicas da NF1: máculas hipercrômicas com bordas regulares, ovaladas, de tamanhos variados, em grande número, com tonalidades diferentes entre pacientes ou mesmo, na mesma pessoa.
Do ponto de vista histológico, considera-se que a diferença de cor das MCL estaria na maior concentração de melanina nos melanócitos e nos queratinócitos e na presença de macromelanossomas. (Shah 2010, De Shepper et al, 2006). Questiona-se se há ou não diferenças histológicas entre as MCL da NF1 das demais MCL.
As MCL não apresentam tendência a malignização e os tratamentos tentados têm apenas objetivo cosmético. (Shah 2010) Vários tipos de laser têm sido tentados para clareamento das manchas, com resultados variáveis: pulsed dye laser, Er:YAG (erbium doped yttrium aluminum garnet), QS Nd:Yag (Q-switched neodymium-dopet yttrium aluminum garnet), QSRL (Q-switched ruby laser) e QSAL (Q-switched alexandrite laser). Um resultado satisfatório ocorre em cerca de 75% dos pacientes com uma taxa de clareamento maior que 50%. O laser QS-1064-nm Nd:YAG parece ser o mais eficaz com poucos efeitos colaterais. (Zi-Zhen 2023)
OUTRAS SÍNDROMES ASSOCIADAS COM MANCHAS CAFÉ COM LEITE
SÍNDROME DE LEGIUS
Na síndrome de Legius há múltiplas MCL em associação com efélides axilares e/ou inguinais, macrocefalia, pectus excavatum ou carinatum, polidactilia, lipomas, dificuldades de aprendizado, comportamento autista, dismorfismos faciais Noonan símile. Mas os pacientes não apresentam nódulos de Lisch, alterações ósseas, neurofibromas ou outros tumores de nervos periféricos como na NF1. A síndrome de Legius é indistinguível da NF1 em relação ao número, morfologia e padrão de distribuição das MCL. (Shah 2010)
SCHWANNOMATOSE (NF TIPO 2)
Embora os pacientes com Schwannomatose apresentem um número menor de MCL quando comparados com os pacientes com NF1, o número ainda é maior que o da população geral. As MCL não estão entre os critérios diagnósticos para a Schwannomatose, mas estão presentes em 33 a 43% dos pacientes. Além dos schwannomas vestibulares que são característicos dessa desordem, meningiomas, schwannomas espinhais e cutâneos podem estar presentes.
MÚLTIPLAS MANCHAS CAFÉ COM LEITE FAMILIARES
MCL múltiplas familiares são observadas em famílias sem outras características de NF1 ou outras síndromes. A ausência de neurofibromas, nódulos de Lisch e outras alterações fenotípicas nos faz pensar neste diagnóstico. Do ponto de vista genético não há mutações nos genes conhecidos das demais síndromes que cursam com MCL. Anteriormente eram categorizadas como NF6 na classificação de Riccardi.
SÍNDROME DE NOONAN
A síndrome de Noonan é uma desordem autossômica recessiva com mutações descritas em vários genes que pertencem a via RAS-MAPK. Caracteriza-se pelas seguintes alterações: baixa estatura, anomalias cardíacas, defeitos no tórax como pectus excavatum ou carinatum, retardo mental, hipertelorismo, ptose palpebral, fendas palpebrais inclinadas para baixo, orelhas com implantação baixa e rodadas posteriormente, fronte proeminente, filtro labial largo com bordas labiais proeminentes, pescoço largo, alterações hemorrágicas, presença de lentigos e manchas café com leite.
SÍNDROME DO CROMOSSOMO EM ANEL
MCL múltiplas tem sido relatadas em pacientes com síndromes do cromossomo em anel envolvendo os cromossomos 7, 11, 12, 15 e 17. Essas crianças tendem a apresentar uma variedade de outras anomalias congênitas, incluindo dismorfismo facial, microcefalia, clinodactilia, baixa estatura e déficits neurocognitivos. A possibilidade de didimose (“Twin spotting”) é sugerido pelo relato de crianças com MCL múltiplas associadas com máculas hipopigmentadas.
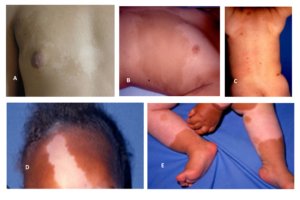
Figura 4: A: Manchas gêmeas (“twin spotting”) – manchas hipocrômicas (nevo despigmentado ou hipopigmentado) associadas com hipercrômicas; B: Mancha café com leite segmentar e terminando na linha média; C, D e E: Piebaldismo.
SÍNDROME MCCUNE-ALBRIGHT
As características principais incluem displasia fibrosa poliostótica, puberdade precoce, outras endocrinopatias como hipertireoidismo, hiperparatireoidismo, síndrome de Cushing, acromegalia, MCL grandes, segmentares e com bordas irregulares. As MCL têm uma predileção para áreas com proeminência óssea (fronte, nuca, tórax, nádegas e região sacral) e geralmente são unilaterais, não ultrapassando a linha média (padrão de mosaicismo em tabuleiro de xadrez).
SÍNDROME DE DEFICIÊNCIA CONSTITUCIONAL DO REPARO DO DNA
A síndrome da deficiência constitucional do reparo do DNA é causada por mutações em genes que fazem o reparo no DNA e caracteriza-se por uma alta prevalência de cânceres sincrônicos e metacrônicos na infância. Várias neoplasias podem ser detectadas antes dos 18 anos de idade: neoplasias hematológicas; carcinomas colorretais, do endométrio, intestino delgado, ureter, pelve renal, trato biliar, estômago, bexiga; tumores embriológicos, entre outros. Esses pacientes podem apresentar MCL, neurofibromas e efélides axilares de forma semelhante aos pacientes com NF1.
PIEBALDISMO
O piebaldismo é uma desordem autossômica dominante que se caracteriza pelas seguintes características fenotípicas: mecha branca frontal nos cabelos, mácula acrômica triangular na fronte, máculas acrômicas bilaterais no tronco e membros e manchas hipercrômicas (MCL) dentro das manchas acrômicas e na pele normal.
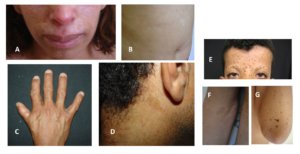
Figura 5 – A e B: Síndrome de Bloom; C e D: Anemia de Fanconi; E, F e G: Síndrome de Leopard.
SÍNDROME DE BLOOM
A síndrome de Bloom é uma doença autossômica recessiva causada por mutação no gene BLM 15q26.1. Caracteriza-se pela tríade: eritema telangiectásico na face, fotossensibilidade e retardo de crescimento. Os pacientes apresentam uma face estreita, pequena, triangular com nariz proeminente (face de passarinho). Podem apresentar também hipogonadismo, imunodeficiências e predisposição para desenvolvimento de malignidades. As MCL podem estar presentes em 50% dos casos e geralmente junto com manchas hipocrômicas, especialmente no tronco.
ANEMIA DE FANCONI
Caracteriza-se por baixa estatura, distúrbios pigmentares na pele (hipermelanose difusa, manchas café com leite, manchas hipocrômicas) presentes em 40% dos casos, malformações ósseas (defeitos no polegar e antebraços), renais, cardíacas, TGI, SNC, hipogonadismo, endocrinopatias. A falência da medula óssea ocorre em 90% dos casos e malignidades não hematológicas podem ocorrer em 20 a 30% dos casos.
SÍNDROME LEOPARD (LENTIGOS MÚLTIPLOS)
LEOPARD é um acrônimo que significa: Lentigos simples; alterações no Eletrocardiograma; hipertelorismo Ocular; cardiomiopatita Obstrutiva; estenose Pulmonar; Anomalias genitais e reprodutivas; Retardo de crescimento; surdez (Deafness).
Trata-se de uma doença autossômica dominante, alélica a síndrome de Noonan, caracterizada pela presença de lentigos que surgem após 4-5 anos de idade, aumentam em número até a puberdade e não acometem as mucosas. As MCL surgem antes dos lentigos e estão presentes em 70 a 80% dos pacientes. Nesta síndrome há também manchas mais escuras e maiores que os lentigos denominadas manchas café noir.
OUTRAS MANIFESTAÇÕES DERMATOLÓGICAS PRESENTES NA NF1 ALÉM DAS MCL
EFÉLIDES NAS DOBRAS CUTÂNEAS (SINAL DE CROWE)
As efélides são a segunda característica mais comum na NF1. Geralmente surgem entre 2 a 3 anos de idade, nas axilas e/ou virilhas, surgindo portanto, mais tardiamente, quando comparadas com as MCL. Cerca de 90% dos adultos com NF1 tem efélides nas axilas e virilhas. Podem ocorrer também no pescoço, mamas, região perioral, proximal dos membros e mesmo tronco, mas nestes locais não são consideradas critério diagnóstico para NF1. Variam de 1 a 3 mm, distinguindo assim, das MCL. (Boyd et al., 2009)(Friedman 2016)
O termo efélides é utilizado para descrever manchas pequenas, hiperpigmentadas encontradas apenas em áreas expostas ao sol, principalmente em indivíduos de pele clara. Contrariando esta descrição, na NF, a mesma denominação é utilizada para manchas de aspecto similar, mas presentes principalmente em áreas de dobras e não expostas ao sol.
Os achados histológicos são idênticos aos das MCL e alguns autores as consideram como MCL pequenas e agrupadas. Quando apenas as MCL e efélides axilares /inguinais estão presentes, o diagnóstico de síndrome de Legius deve ser considerado.
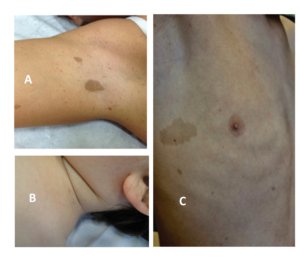
Figura 6 – A e B: Manchas café com leite e efélides axilares e cervicais; C: disseminadas.
HIPERPIGMENTAÇÃO DIFUSA GENERALIZADA
Uma hiperpigmentação generalizada é encontrada nos pacientes com NF1 quando comparados com pais e irmãos não acometidos. Uma hipótese para explicar esta alteração é que essa hipercromia seria pela mutação nas células germinativas e as MCL seriam secundárias a pelo menos uma segunda mutação (mutações nas células germinativas e somáticas). Nos pacientes com neurofibromatose segmentar pode haver uma hiperpigmentação de base no seguimento anatômico acometido. (Boyd 2009)
NEUROFIBROMAS
Os neurofibromas são outro sinal marcante da NF1. Podem ocorrer em qualquer lugar do corpo e apresentam grande variação na forma e tamanho. (Boyd 2009) Os neurofibromas dérmicos geralmente surgem na adolescência e raramente são detectados na infância.
Há 3 tipos histológicos de neurofibromas: endoneurais, perineurais e epineurais e sua distinção é importante pois sua evolução natural é bastante diferente. Os axônios com sua bainha de mielina se agrupam formando feixes interligados por uma membrana de tecido conjuntivo chamada endoneuro. Em volta destes feixes, há o perineuro. E vários feixes são cobertos por uma membrana mais externa, denominada epineuro.
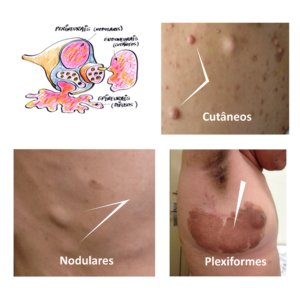
Figura 7 – Diagrama dos diferentes tipos de neurofibromas e fotos correspondentes.
Neurofibromas cutâneos
Neurofibromas endoneurais (cutâneos ou dérmicos ) derivam do endoneuro dos nervos sensitivos terminais cutâneos e são constituídos de células de Schwann, fibroblastos, células endoteliais, perícitos e mastócitos. Podem ser sésseis, achatados ou pedunculados; macios ou com a consistência de borracha; cor da pele a ligeiramente hipercrômicos; tamanhos variados (alguns mm a vários cm). Podem ser dolorosos e/ou pruriginosos. Geralmente surgem na puberdade e podem aumentar de tamanho e número na vida adulta. Além da puberdade, a gravidez é outro período que propicia aumento no número e tamanho dos neurofibromas. (Friedmann 2016)
Os neurofibromas cutâneos provocam grandes problemas para os pacientes com NF1, dependendo da visibilidade, do número e tamanho desses tumores. (Boyd 2009) A excisão cirúrgica com fechamento primário pode ser realizada. A eletrodissecção é uma técnica que permite retirar um número maior de lesões. (Lutterodt 2016)
Neurofibromas subcutâneos
Neurofibromas subcutâneos são revestidos pelo perineuro. Podem formar cordões com vários neurofibromas ao longo do trajeto de um nervo, como contas de um rosário. Apresentam consistência mais endurecida que os neurofibromas cutâneos. Geralmente surgem nos primeiros 5 anos de vida portanto, mais precocemente quando comparados com os neurofibromas cutâneos. Podem crescer ao longo da vida, causar desconforto como dor e/ou prurido e sofrer transformação maligna.
Neurofibromas plexiformes
Os neurofibromas plexiformes (epineurais) ocorrem em cerca de 30-50% dos pacientes com NF1. (Hirbe 2014) São congênitos, o que ajuda diferenciá-los dos neurofibromas cutâneos e subcutâneos. Apresentam pele sobrejacente hipercrômica, com ou sem hipertricose, podendo ser confundido com uma MCL ou nevo melanocítico congênito. (Hernández-Martin 2016). Crescem principalmente nas primeiras 3 décadas de vida no trajeto de um nervo. Podem ser muito grandes e pesados, desfigurantes e interferirem com a função da área acometida. A malignização dos tumores neurais da NF1 usualmente ocorrem em neurofibromas plexiformes, em cerca de 8 a 15% dos pacientes. Dor e crescimento rápido destes tumores é um sinal de alerta importante. A malignização geralmente ocorre na adolescência ou vida adulta. Os neurofibromas plexiformes podem ser internos. (Boyd 2009, Riccardi 2016, Friedman 2016)
NEUROFIBROMAS NA CAVIDADE ORAL
Neurofibromas na cavidade oral ocorrem em 4-7% dos pacientes com NF1 e se apresentam como tumores de tamanhos variados, não dolorosos, localizados principalmente na língua, palato, mucosa bucal e alveolar e assoalho da boca. Quando traumatizados, podem causar sintomas. Exérese cirúrgica, eletrocauterização ou laser diodo ou erbium podem ser realizados. (Angiero 2016)
MÁCULAS PSEUDOATRÓFICAS E AZUL AVERMELHADAS
São variantes incomuns dos neurofibromas. A mácula azul avermelhada foi descrita como uma lesão de limites mal definidos, consistência macia, localizada no tronco. Surgem um pouco antes ou durante a adolescência e ficam levemente elevadas com o tempo. Histologicamente, caracterizam-se pela presença de vasos com parede espessada e lúmen largo e células neurais na derme papilar e reticular. (Boyd 2009)
As máculas pseudoatróficas são lesões ovais, levemente deprimidas, medindo 5 a 10 cm. A cor e textura são semelhantes às da pele normal, mas quando comprimidas, demonstram uma perda do tecido subcutâneo subjacente. O exame histológico demonstrou redução do colágeno na derme reticular e substituição por tecido neuroide composto por células de Schwann, fibroblastos e vasos (hipoplasia dérmica neurofibromatosa). (Boyd 2009)
XANTOGRANULOMA JUVENIL
A tripla associação, NF1, xantogranuloma juvenil e leucemia mielomonocítica, é questão de debate. Há relatos na literatura demonstrando uma prevalência de xantogranuloma juvenil em 9 a 30% dos pacientes com NF1. Esta prevalência muda com a idade, porque os xantogranulomas podem já ter regredido na época em que o diagnóstico de NF1 é definido. A maioria dos xantogranulomas regride antes dos 5 anos de idade. (Jans 2015; Fenot 2014). O risco de desenvolver leucemia mieloide mielomonocítica juvenil é 200 a 500 vezes maior em crianças com NF1. A leucemia mieloide mielomonocítica juvenil surge habitualmente antes dos 6 anos de idade, portanto deve ser pensada em crianças que tenham sinais sugestivos ou diagnósticos de NF1 e lesões de xantogranuloma juvenil. (Jans 2015) (Boyd 2009)
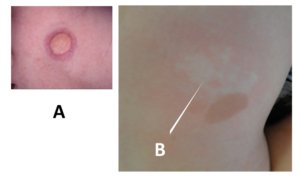
Figura 8. Outras lesões cutâneas que podem ser encontradas nas pessoas com NF1. A: nódulos amarelo-hipercrômicos, um xantogranuloma juvenil; B: nevo anêmico.
OUTROS TUMORES
Postula-se uma maior chance dos pacientes com NF1 em ter tumor glômico. O reconhecimento é importante por ser um tumor doloroso e a exérese cirúrgica é curativa. (Boyd 2009) Rabdomiossarcoma, feocromocitoma, tumores no trato gastrintestinal, câncer de mama em mulheres abaixo de 50 anos são mais frequentes em pacientes com NF1. (Friedman 2016) Em relação ao melanoma, a relação é controversa. A presença de melanomas mais espessos em pacientes com NF1 pode ser pela grande quantidade de lesões pigmentadas nestes pacientes, o que pode ocasionar demora no diagnóstico. (Boyd 2009) Lipomas também ocorrem mais frequentemente em pacientes com NF1.
NEVO ANÊMICO
Embora a primeira descrição da associação entre nevo anêmico e NF1 ter sido feita por Naegeli em 1915 e novamente relatada por outros autores em 1969 e 1970, essa associação recebeu pouca atenção até há poucos anos. (Hernández-Martín 2015) A partir de 2013, séries com grande número de pacientes com NF1 e nevo anêmico foram publicadas, demonstrando uma prevalência dessa associação em cerca de 50% dos pacientes. O nevo anêmico encontra-se especialmente no tronco, principalmente na região anterior da parede torácica e esternal, mas pode estar presente em qualquer local do corpo. O tamanho é variável, sendo frequentemente múltiplo. Anormalidades vasculares tem sido descritas em pacientes com NF1, principalmente nas artérias renais e cerebrais. (Hernández-Martín et al., 2015) A presença do nevo anêmico pode ser útil para diferenciar NF1 de outras genodermatoses que apresentam MCL e efélides.
PRURIDO
O prurido é uma manifestação relativamente comum na NF1, estimado ser uma queixa em cerca de 20% dos pacientes. (Brenaut 2016). Quando generalizado, parece estar associado a um maior número de mastócitos nos neurofibromas. Pode ser localizado, considerado como uma causa de prurido neuropático, por tumores no sistema nervoso central (medular ou encefálico). Geralmente está associado a outros sintomas como dor, alodinia, parestesias, hiperestesias ou sensações de choque. (Boyd 2009; Brenaut 2016)
Referências:
Antônio JR, Goloni-Bertollo EM, Trídico LA. Neurofibromatosis: chronological history and current issues. An Bras Dematol. 2013; 88(3):329-43.
Friedman JM. Initial posting: October 2, 1998; last update May 17, 2018. Pagon RA, Adam MP, Arlinger HH, et al., editors. GeneReviews [internet]. Seatle (WA): University of Washington, Seatle; 1993-2018. http://www.ncbi.nlm.nih.gov/books/NBK/1109/
Nunley KS, Gao F, Alber AC, Bayliss SJ, Gutmann DH. Predictive value of café au lait macules at initial consultation in the diagnosis of neurofibromatosis type 1. Arch Dermatol. 2009; 145(8):883-7.
Abdolrahimzadeh B, Piraino DC, Albanese G, Cruciani F, Rahimi S. Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography. Clin Ophthalmol. 2016;10:851-60.
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000; 105:608-14.
Pereira LB. Prevalência de dermatoses no recém-nascido: estudo comparativo entre dois hospitais de Belo Horizonte, Brasil. Tese. Belo Horizonte: Universidade Federal de Minas Gerais, 1997.
Korf BR. Diagnostic outcome in children with multiple café au lait spots. Pediatrics 1992; 90(6):924-7.
Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014; 13:834-43.
Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009; 61:1-14.
Boyd KP, Gao L, Feng R, Beasley M, Messiaen L, Korf BR et al.. Phenotypic variability among café-au-lait macules in neurofibromatosis type 1. J Am Acad Dermatol. 2010; 63:440-7.
Riccardi VM. NF1 Clinical elements and the NF1 neurofibroma burden. JJ Neur Neurrosci. 2016; 3(1):025.
Rodriguez-Jimenez P, Chicharro P, Munõz E, Dauden E. Long-term treatment of neurofibromatosis 1 with ketotifen. A report of three cases. Am J Med Genet A. 2016; 170A(4):1092-4.
Duman N, Elmas M. Dermoscopy of cutaneous neurofibromas associated with neurofibromatosis type 1. J Am Acad Dermatol. 2015; 73(3):529-31.
Lutterodt CG, Mohan A, Kirkpatrick N. The use of electrodessication in the treatment of cutaneous neurofibromatosis: a retrospective patient satisfaction outcome assessment. J Plast Reconstr Aesthet Surg. 2016; 69(6):765-9.
Angiero F, Ferrante F, Ottonello A, Maltagliati A, Crippa R. Neurofibromas of the oral cavity: clinical aspects, treatment, and outcome. Photomed Laser Surg. 2016; 34(2):56-60.
Hernández-Martín A, Duat-Rodríguez A. An update on neurofibromatosis type 1: not just café-au-lait spots, freckling, and neurofibromas. An update. Part I. Dermatological clinical criteria diagnostic of the disease. Actas Dermosifiliogr. 2016; 107(6):454-64.
Jans SRR, Schomerus E, Bygum A. Neurofibromatosis type 1 diagnosed in a child based on multiple juvenile xanthogranulomas and juvenile myelomonocytic leukemia. Pediatr Dermatol. 2015; 32(1): e29-32.
Fenot M, Stalder JF, Barbarot S. Juvenile xanthogranulomas are highly prevalent but transient in young children with neurofibromatosis type 1. J Am Acad Dermatol. 2014; 71(2):389-90.
De Schepper S, Boucneau J, Haeghen YV, Messiaen L, Naeyaert J, Lambert J. Café-au-lait spots in neurofibromatosis type 1 and in healthy control individuals: hyperpigmentation of a different kind? Arch Dermatol Res. 2006; 297:439-49.
Hernández-Martín A, García-Martínez FJ, Duat A, López-Martín I, Noguera-Morel L, Torrelo A. Nevus anemicus: a distinctive cutaneous finding in neurofibromatosis type 1. Pediatr Dermatol. 2015; 32(3):342-7.
Brems H, Legius E. Legius syndrome, an update. Molecular pathology of mutations in SPRED1. Keio J Med. 2013; 62(4):107-12.
Benelli E, Bruno I, Belcaro C, Ventura A, Berti I. Legius syndrome: case report and review of literature. Ital J Pediatr. 2015; 41:8.
Polder KD, Landau JM, Vergilis-Kalner IJ, Goldberg LH, Friedman PM, Bruce S. Laser eradication of pigmented lesions: a review. Dermatol Surg. 2011; 37 (5):572-95.
Brenaut E, Nizery-Guermeur C, Audebert-Bellanger S, Ferkal S, Wolkenstein P, Misery L et al. Clinical characteristics of pruritus in neurofibromatosis 1. Acta Derm Venereol. 2016;96:398-9.
Shah KN. The diagnostic and clinical significance of café-au-lait macules. Pediatr Clin N Am. 2010; 57:1131-53.
Pereira LB, Gontijo B. Dermatoses neonatais de importância clínica: notificação no prontuário do recém-nascido. J Pediatr. 1999; 75(5):357-60.
Kehrer-Sawatzki H, Cooper DN. Challenges in the diagnosis of neurofibromatosis type 1 (NF1) in young children facilitated by means of revised diagnostic criteria including genetic testing for pathogenic NF1 gene variants. Hum Genet. 2022; 141:177–91.
Torrelo A, Baselga E, Nagore E, Zambrano A, Happle R. Delineation of the various shapes and patterns of nevi . Eur J Dermatol 2005; 15 (6): 439-50.
Zi-Zhen G, Zhi-Cho W, Dun W, Lin-Ling G, Yue-Hua L, Yi-Hui G, Wei W et al. Laser treatment for café-au-lait macules: a systematic review and meta-analysis. Eur J Med Res.2023; 28:185.