Um grupo de cientistas do Centro de Referência em Neurofibromatoses, do Hospital das Clínicas da Universidade Federal de Minas Gerais, descobriu que mulheres com neurofibromatose do tipo 1 (NF1) apresentam aumento do metabolismo corporal em repouso, ou seja, o seu consumo de energia está aumentado em relação às pessoas sem a NF1 do mesmo sexo, idade, estatura e peso.
Junto com este metabolismo aumentado, elas têm menor estatura, redução da massa muscular e força muscular diminuída, concordando com estudos anteriores realizados em nosso Centro (VER AQUI )
As causas deste aumento do metabolismo nas mulheres com NF1 ainda estão sendo investigadas. Uma das explicações prováveis seria a ativação exagerada de certas vias metabólicas dentro das células (via RAS), por causa da deficiência de neurofibromina causada pela mutação no gene NF1.
A importância destes resultados está na melhor compreensão do desenvolvimento corporal das pessoas com NF1, uma linha de pesquisa iniciada por este grupo de cientistas, o que poderá, no futuro, contribuir para novos tratamentos da neurofibromatose do tipo 1.
O artigo científico foi publicado na revista internacional “Clinical Nutrition” (Nutrição Clínica) e recebeu o título “Increased resting metabolism in neurofibromatosis type 1” (Aumento do metabolismo de repouso na neurofibromatose do tipo 1) ( ver aqui o link para o artigo completo ).
Até o dia 6 de agosto de 2019 o artigo completo pode ser baixado deste link: CLIQUE AQUI
Os autores e autoras da pesquisa foram Márcio Leandro Ribeiro de Souza, Ann Kristine Jansen, Luiz Oswaldo Carneiro Rodrigues, Darlene Larissa de Souza Vilela, Adriana Maria Kakehasi, Aline Stangherlin Martins, Juliana Ferreira de Souza e Nilton Alves de Rezende.
A equipe científica recebeu apoio financeiro de três agências públicas de financiamento: CAPES, CNPq e FAPEMIG.
Abaixo a tradução do resumo
Neurofibromatose tipo 1 (NF1) é uma doença de genética autossômica dominante que é caracterizada por alterações neurocutâneas com envolvimento multissistêmico. Um estudo anterior com adultos com NF1 revelou que mudanças no gasto de energia total estavam relacionadas com a composição corporal e consumo de alimentos. O gasto de energia de repouso (REE), uma medida de energia que o corpo gasta para manter as funções vitais, ainda não foi avaliado em populações de NF1. Este estudo objetivou avaliar REE em indivíduos com NF1 utilizando calorimetria indireta (IC) e avaliar sua correlação com a força muscular e a composição do corpo.
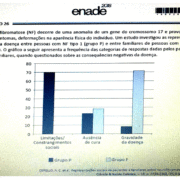
Vinte e seis adultos com NF1 (14 homens) de idade entre 18 – 45 anos submeteram-se a IC para avaliar REE, quociente respiratório (RQ) e a utilização de substrato. A composição corporal foi avaliada por absorção de raio-x de dupla energia. Peso, altura e circunferência da cintura (WC) também foram medidos. Força muscular máxima (Smax) foi mensurada pelo teste de apreensão usando um dinamômetro. Os pacientes do grupo de NF1 foram comparados com 26 controles saudáveis no grupo controle, que foram combinados por sexo, idade, índice de massa corporal (IMC) e nível de atividade física.
Não houve diferença no peso, WC, massa gorda e percentual de gordura corporal (BFP). Apendicular massa magra (ALM) ajustado pelo IMC (ALMBMI) (0.828 ± 0.161 a versus 0.743 ± 0,190; P = 0,048) e Smax (37,5 ± 10,6 a versus ± 31,1 12.2; P = 0,035) foi menor no grupo de NF1 do que no grupo controle. Não há diferenças na composição corporal, força e parâmetros antropométricos foram observadas em homens, mas mulheres com NF1 apresentaram a menor área de superfície corporal (BSA), massa corporal magra (MCM), ALM, ALMBMI e Smax. REE ajustada pelo peso, LBM ou ALM foi maior no grupo de NF1 do que no grupo controle (medianas, 21,9 contra 26,3, P = 0,046; 36,5 versus 41.1, P = 0,012; e 82,3 contra 92,4, P = 0,006, respectivamente), e essas diferenças foram observadas somente entre as mulheres. RQ foi menor no grupo de NF1 do que no grupo controle (0,9 ± 0,1 a versus 0,8 ± 0,1; P = 0,008), revelando que os indivíduos com NF1 oxidado mais lípidos e hidratos de carbono menos do que controles. REE correlação negativa com o BFP e positivamente com peso, estatura, BMI, WC, BSA, LBM, ALM, ALMBMI, densidade óssea mineral e Smax.
Conclusão
Indivíduos com NF1, particularmente as mulheres, apresentaram aumento do REE (ajustado pelo peso, LBM ou ALM) e redução do RQ em comparação com controles saudáveis. Esses achados estavam associados com baixa ALMBMI e Smax, indicando possivelmente sarcopenia prematura nesta população. Investigação adicional sobre metabolismo energético em NF1 e diferenças de gênero pode ser útil para explicar os mecanismos destas alterações.
Abaixo o resumo em inglês.
Background & aims
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disease that is characterized by neurocutaneous changes with multisystem involvement. A previous study with adults with NF1 revealed that changes in total energy expenditure were related to food consumption and body composition. Resting energy expenditure (REE), a measure of energy that the body expends to maintain vital functions, has not been assessed in NF1 populations. This study aimed to assess REE in individuals with NF1 using indirect calorimetry (IC) and evaluate its correlation with body composition and muscle strength.
Methods
Twenty-six adults with NF1 (14 men) aged 18–45 years underwent IC for assessing REE, respiratory quotient (RQ), and substrate utilization. Body composition was assessed by dual energy X-ray absorptiometry. Weight, height, and waist circumference (WC) were also measured. Maximum muscular strength (Smax) was measured by handgrip test using a dynamometer. Patients in the NF1 group were compared to 26 healthy controls in the control group, who were matched by sex, age, body mass index (BMI), and physical activity level.
Results
There were no differences in weight, WC, fat mass, and body fat percentage (BFP). Appendicular lean mass (ALM) adjusted by BMI (ALMBMI) (0.828 ± 0.161 versus 0.743 ± 0.190; P = 0.048) and Smax (37.5 ± 10.6 versus 31.1 ± 12.2; P = 0.035) was lower in the NF1 group than in the control group. No differences in body composition, strength, and anthropometric parameters were observed in men, but women with NF1 presented lower body surface area (BSA), lean body mass (LBM), ALM, ALMBMI, and Smax. REE adjusted by weight, LBM, or ALM was higher in the NF1 group than in the control group (medians, 21.9 versus 26.3, P = 0.046; 36.5 versus 41.1, P = 0.012; and 82.3 versus 92.4, P = 0.006, respectively), and these differences were observed only among women. RQ was lower in the NF1 group than in the control group (0.9 ± 0.1 versus 0.8 ± 0.1; P = 0.008), revealing that individuals with NF1 oxidized more lipids and fewer carbohydrates than controls. REE correlated negatively with BFP and positively with weight, height, BMI, WC, BSA, LBM, ALM, ALMBMI, bone mineral content, and Smax.
Conclusions
Individuals with NF1, particularly women, presented with increased REE (adjusted by weight, LBM, or ALM) and lower RQ compared to healthy controls. These findings were associated with lower ALMBMI and Smax, possibly indicating premature sarcopenia in this population. Further investigation concerning energy metabolism in NF1 and gender differences may be helpful in explaining underlying mechanisms of these changes.