Pergunta da leitora E.M.L. de Natal, RN: ”Minha filha com 24 anos estava bem até 6 meses atrás quando sentiu uma dor forte na barriga e no pronto atendimento do SUS foi feito ultrassom que suspeitou de tumor. Fizeram ressonância e apareceram tumores nas raízes dos nervos. A dor melhorou , mas a neurologista pediu ressonância completa que mostrou tumores em toda a coluna. Ela disse que é neurofibromatose do tipo 1. Achei a página da AMANF na internet e estou com dúvida. Minha filha tem apenas uma mancha café com leite, não tem sardinhas debaixo do braço, não tem dificuldade de aprendizado e nem neurofibromas cutâneos. Como pode ser NF1?”
Cara E,
Obrigado pela sua pergunta, pois ela poderá ser útil a outras famílias. O que você descreveu junto com o laudo da ressonância que nos enviou, indicam que sua filha apresenta uma forma especial e mais rara da NF1, chamada neurofibromatose espinhal, descrita inicialmente pelo nosso querido e saudoso amigo Dr. Vincent M. Riccardi (1991).
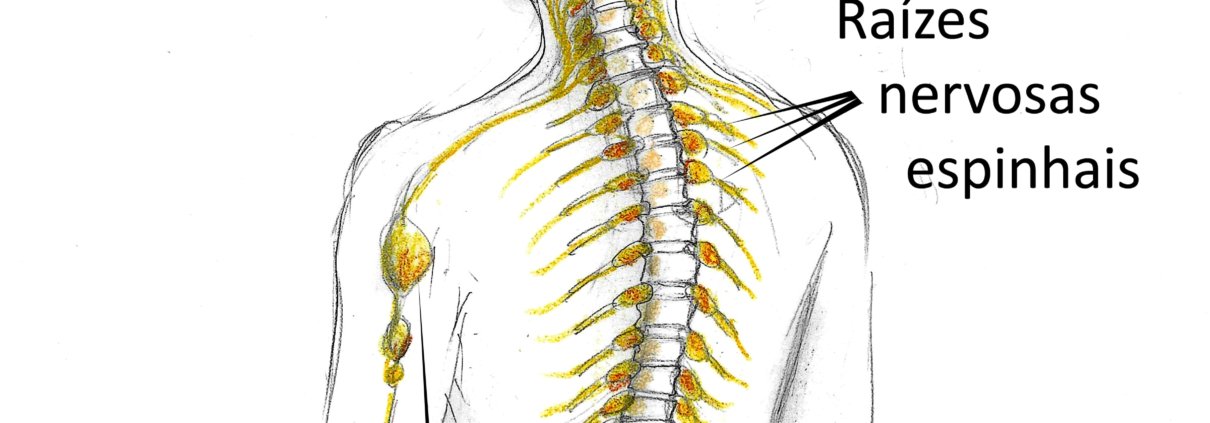
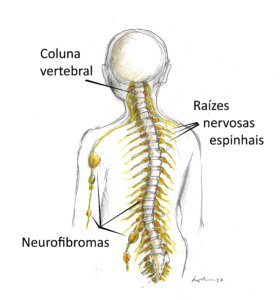
Essa forma espinhal da NF1 se caracteriza pela presença de neurofibromas em TODAS as raízes dos nervos espinhais, de ambos os lados, desde a coluna cervical até a coluna lombossacral. Também pode haver neurofibromas nos nervos periféricos, formando estruturas parecidas com um colar de contas debaixo da pele (ver figura ilustrativa acima).
O diagnóstico da forma espinhal necessita da confirmação de neurofibromas em todas as raízes espinhais por meio da realização da ressonância magnética da coluna vertebral.
A forma espinhal da NF1 ocorre em cerca de 2% de todas as pessoas com NF1, ou seja, é rara. Em nosso ambulatório no Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais (CRNF), temos diagnosticado a forma espinhal em cerca de 0,5% das pessoas atendidas, talvez porque nossa população realiza menos exames de imagem (como ressonância magnética) da coluna vertebral.
As pessoas com a forma espinhal apresentam menos sinais e sintomas da forma mais comum da NF1, como acontece com sua filha.
Geralmente elas têm poucas manchas café com leite, as quais são maiores, mais claras e de bordas irregulares. Além disso, elas apresentam poucos neurofibromas cutâneos (ou nenhum) e geralmente não têm dificuldades cognitivas ou escolares.
Tudo isso, dificulta e atrasa o diagnóstico de muitas pessoas com a forma espinhal da NF1.
Apesar de ser uma forma mais rara da NF1, compreender seu funcionamento nos ajuda a tratar melhor as pessoas acometidas e a entender a NF1 de um modo geral.
Quais são os sinais e sintomas da forma espinhal da NF1?
A partir de duas revisões científicas sobre a forma espinhal da NF1 (ver aqui a revisão de 2015 com 49 casos e ver aqui a revisão de 2023 com 98 casos), assim como da nossa experiência com dezessete pessoas com a forma espinhal diagnosticada nos últimos dez anos, podemos reconhecer algumas características dessa forma mais rara da NF1, comparada com a forma clássica (ver aqui revisão de 2025):
- Principal: neurofibromas ou espessamentos de TODAS as raízes nervosas espinhais com alargamento dos forames de conjugação vertebrais (ressonância magnética); Às vezes, encontramos espessamento de todos os nervos e não as formações ovaladas típicas dos neurofibromas.
- Painel genético com variante patogênica no gene NF1 (especialmente se for do tipo missense – ver abaixo).
- Neurofibromas subcutâneos nodulares ou profundos nos nervos periféricos (frequentes);
A frequência das demais características da NF1 são apresentadas na Tabela abaixo, na qual comparamos a forma clássica da NF1 com a forma espinhal num estudo italiano e com a nossa experiência no CRNF.
| NF1 | |||
| Clássica | Espinhal | ||
| Características | Friedmann | Ruggieri
49 casos |
CRNF
17 casos |
| História familiar | 50% | 55% | 35% |
| Manchas café com leite | 99% | 67% | 94% |
| Efélides | 85% | 18% | 59% |
| Nódulos de Lisch | 95% | 24% | 25% |
| Alterações da coroide | 90% | # | # |
| Glioma da via óptica | 18% | 10% | 18% |
| Glioma não óptico | 4% | 4% | # |
| Neurofibromas | |||
| Cutâneos | 99% | 24% | 41% |
| Nodulares subcutâneos | 15% | # | 76% |
| Plexiformes (difusos) | 30% | 6% | 0% |
| Tumor maligno | 10% | 6% | 13% |
| Deficiência intelectual | 6% | # | 0% |
| Dificuldades de aprendizagem | 55% | 7% | 0% |
| Problemas de comportamento | 40% | # | 0% |
| Convulsões | 7% | # | 6% |
| Displasia de ossos longos | 2% | rara | 6% |
| Escoliose distrófica | 5% | # | 0% |
| Escoliose não distrófica | 5% | 18% | # |
| Osteoporose | 20% | # | # |
| Hipertensão | 20% | # | 0% |
| Baixa estatura e macrocrania | 50% | raras | 0% |
#: dados não medidos ou não relatados
Em conclusão, a forma espinhal da NF1 é bastante diferente da NF1 clássica.
Quais os sintomas da NF1 espinhal?
Os neurofibromas das raízes espinhais geralmente estão presentes muito cedo na infância, mas podem permanecer sem sintomas até mais tarde na vida (20 a 30 anos) e alguns casos continuam assintomáticos até os cinquenta anos.
Algumas vezes a NF1 na forma espinhal é descoberta por acaso, ao se fazer uma ressonância da coluna por qualquer outro motivo.
Em algumas crianças aparece um neurofibroma nodular solitário periférico debaixo da pele, que chamamos de “sentinela”, o qual pode sugerir a forma espinhal.
Os homens com a forma espinhal costumam apresentar sintomas mais cedo (em torno dos 16 anos) do que as mulheres (em torno dos 26 anos), ao contrário da forma clássica, na qual não há diferença para início dos sintomas entre homens e mulheres.
Os sintomas podem se iniciar em um lado do corpo ou em ambos os lados.
Os principais sintomas são:
- Déficits sensoriais: podem surgir dormência, formigamento, sensibilidade alterada, choques.
- Déficits motores: podem surgir redução ou perda da força (paresia) e da coordenação muscular, que podem progredir para a paralisia.
- Dor do tipo neuropática: pode surgir dor forte e muitas vezes requerer a participação de clínicas especializadas no tratamento da dor (ver aqui mais informações sobre este tipo de dor).
Como evolui a forma espinhal ao longo da vida?
O início dos sintomas varia muito de uma pessoa para outra, em algumas na infância, mas na maioria ocorre em torno da segunda década, mas em outras muito mais tarde na vida.
Uma vez iniciados os sintomas, geralmente eles se agravam com o passar do tempo, mas algumas pessoas podem permanecer assintomáticas durante décadas.
Quais os tratamentos para a forma espinhal?
O reconhecimento da forma espinhal é relativamente recente entre os especialistas e, como ela é mais rara do que a forma clássica, ainda não temos estudos científicos sobre tratamentos direcionados especificamente para a forma espinhal.
Mesmo para a NF1 clássica, por enquanto, não há um tratamento capaz de curar por completo a doença, embora diversos estudos científicos estejam sendo realizados em todo o mundo em busca de tratamentos efetivos e esperamos que eles possam ajudar no tratamento da forma espinhal.
Por enquanto, sugerimos:
- As pessoas assintomáticas com a NF1 espinhal precisam de acompanhamento clínico anual, com as medidas preventivas habituais, com atenção especial para a medida da pressão arterial, prevenção de câncer de mama e possíveis sinais de transformação maligna de algum neurofibroma.
- Quando houver sintomas, tenta-se o tratamento clínico, se possível, com medicamentos para as alterações sensoriais e dor e fisioterapia para o fortalecimento muscular e manutenção da capacidade funcional.
- O tratamento cirúrgico pode ser considerado, mas em geral é muito difícil encontrar um ou alguns neurofibromas que sejam a causa evidente dos sintomas. Os estudos de imagem e a eletroneuromiografia podem ajudar a localizar o neurofibroma responsável pelos sintomas e, se algum for identificado, a neurocirurgia pode ser avaliada.
- Nos casos sintomáticos, nos quais não é possível a cirurgia, deve ser considerada a indicação dos medicamentos inibidores MEK (selumetinibe ou mirdametinibe – ver aqui mais informações sobre esta classe de medicamentos). No entanto, até o momento, desconhecemos qualquer estudo científico com os inibidores MEK especificamente para a forma espinhal da NF1.
Os estudos que conhecemos até o momento analisaram o efeito destes medicamentos em neurofibromas nodulares e difusos (chamados plexiformes) que incluíram pessoas com neurofibromas envolvendo as raízes nervosas espinhais, mas não houve a identificação exata se eram pessoas com a forma espinhal da NF1 ou a forma clássica.
Em conclusão, sabendo que a forma espinhal NF1 é diferente da NF1 clássica, ainda não podemos dizer se as pessoas com a forma espinhal respondem do mesmo jeito ao tratamento com inibidores MEK, por isso precisamos de mais estudos neste sentido.
Temos grande expectativa de que num futuro breve teremos medicamentos eficazes para o tratamento da forma espinhal da NF1.
Equipe médica do CRNF HC UFMG
Abril de 2026


Pergunta do leitor S.M.N. – “Encontrei uma informação (ver aqui) sobre pesquisadores do Instituto Nacional de Câncer, da Fiocruz e de outras instituições científicas brasileiras que trabalham para incorporar a terapia com células CAR-T ao SUS. Será que essa técnica pode ser útil para curar a NF1?”
Caro S, muito obrigado pela sua pergunta, pois muitas pessoas nos enviam questões semelhantes sobre novas terapias que vão surgindo em estudos científicos: quando chegará a vez da cura da NF1 e das Schwannomatoses?
Lemos as informações que nos enviou sobre este novo tratamento para tratar alguns tipos de câncer, mas ele não tem relação com a NF1 e nem com a Schwannomatose.
No entanto, nossa esperança de cura para a NF1 e Schwannomatoses é cada vez maior por causa do conhecimento científico que vem sendo construído por pessoas dedicadas nas universidades de todo o mundo. Este conhecimento está crescendo muito rapidamente.
Veja, por exemplo, que na década de 80 ainda não conhecíamos o gene NF1, que atualmente já sabemos que causa a Neurofibromatose do tipo 1quando ele surgem variantes patogênicas (mutações no DNA que atrapalham o funcionamento da proteína neurofibromina).
A Figura abaixo mostra a imagem que foi apresentada para o gene NF1, localizado no cromossomo 17 (parte A), quando, em 1990, ficamos sabendo que pessoas com mutações naquela área do cromossomo apresentavam a neurofibromatose do tipo 1.
Em 2023, ou seja, 33 anos depois, nossa imagem do gene NF1 se tornou muito mais profunda e complexa, como mostra a parte B da Figura abaixo.

Vejam quanto conhecimento já foi construído: cada sigla, cada letra, cada número nessa figura tem um significado científico que têm importância clínica.
Por exemplo, hoje já sabemos que quando um tipo de variante (chamada missense) ocorre na região marcada em azul há uma chance maior da pessoa desenvolver neurofibromas nas raízes dos nervos na coluna vertebral (forma espinhal da NF1). Este conhecimento, quem sabe, um dia poderá ser útil no tratamento, prevenção ou cura destes neurofibromas.
Diversas outras informações clínicas já foram relacionadas com determinadas variantes no gene NF1. Muitas ainda estão em estudo, mas algumas variantes já conhecidas são exemplificadas nos círculos coloridos na Figura acima.
Roxo: casos mais graves, que exigem mais cuidados
Verde: casos menos graves, sem neurofibromas
Laranja: somente manchas café com leite
Azul: com estenose pulmonar
Marrom: aumento dos neurofibromas nodulares e difusos (plexiformes)
Magenta: sem neurofibromas, mas com dificuldades cognitivas
Em conclusão, é assim, através do trabalho coletivo, paciente e rigoroso das pessoas que se dedicam à ciência, que um dia poderemos dispor de tratamentos eficientes para curar, ou pelo menos diminuir, o sofrimento das pessoas com NF1 e Schwannomatoses.
Que este dia chegue o mais breve possível!
“Meu filho de 9 anos apresenta NF1 e escoliose. O que devo fazer?” JBS, de Natal, RN.
Cara J., obrigado pela sua pergunta, pois a escoliose é uma das alterações no desenvolvimento ósseo que ocorrem nas pessoas com NF1 com maior frequência (entre 10 e 60%) do que na população em geral (1 a 3%).
Escoliose significa qualquer curvatura da coluna maior do que 10 graus numa escala própria chamada de Escala de Cobb.
Geralmente, nas crianças com NF1, a escoliose aparece antes dos 14 anos de idade, mas raramente depois. Além disso, costuma se estabilizar ao longo da adolescência.
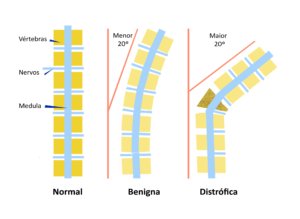
Quando a escoliose é simples, ou seja, apenas uma curvatura suave na coluna e o ângulo for menor do que 20 graus e não encontramos outras alterações ósseas, a evolução geralmente é benigna e bastam exercícios regulares, cuidados com a Vitamina D e observação clínica.
Porém, quando a escoliose é mais acentuada, ou seja, o ângulo da curvatura for maior do que 20 graus e, principalmente, se houver outras alterações ósseas, chamamos de escoliose distrófica, que apresenta maior chance de progressão e necessidade de tratamento cirúrgico.
Sintomas
Os sintomas mais comuns na escoliose são dor em cerca de 20% das pessoas com NF1. Geralmente, a dor ocorre na coluna torácica (10%) e lombar (9%). Pode haver perda da função sensorial (5%) e motora (10%, mais comum no sexo masculino).
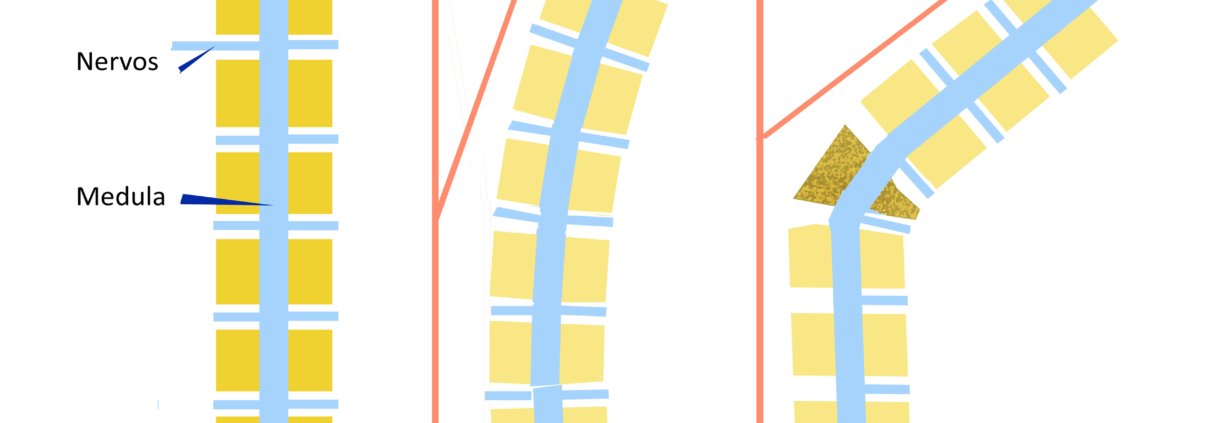
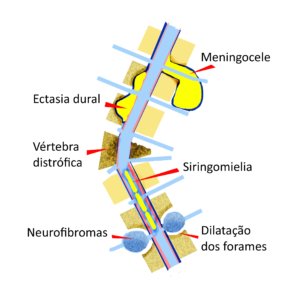
Alterações ósseas associadas com a NF1
Nas pessoas com NF1, algumas alterações ósseas (ver figura) associadas à escoliose indicam a necessidade de maior cuidado médico :
- Deformidades das vértebras (cerca de 5%)
Podem ocorrer distrofias, ou seja, defeitos de formação, quando uma ou mais vértebras apresentam problemas estruturais como desabamento, fraturas, baixa densidade óssea, deformidades e falhas.
- Ectasia dural (45%)
Ectasia quer dizer “fora do lugar” e ocorre quando as membranas que envolvem a medula (chamadas de dura máter e aracnoide) apresentam dilatações e invadem o corpo das vértebras. Se aumentarem ao longo do tempo, precisam ser tratadas para evitar a progressão da escoliose.
- Erosões (20%)
Geralmente associadas com a ectasia dural, são defeitos de formação na parte central das vértebras (chamados de scalloping na ressonância magnética). É uma das alterações mais importantes para o agravamento da escoliose.
- Meningoceles (6%)
Quando as membranas que revestem a medula (dura máter e aracnoide) apresentam dilatações entre os espaços vertebrais, formando bolsas que contém o líquor. Geralmente localizadas na coluna torácica.
- Dilatação dos forames neurais (40%)
Os nervos que saem e entram na coluna vertebral de ambos os lados passam por orifícios chamados forames intervertebrais. Quando surgem neurofibromas nestes nervos, o crescimento dos neurofibromas vai dilatando os forames lentamente e eles se apresentam alargados na ressonância magnética. Geralmente localizadas na coluna lombar e relacionadas com dor.
Siringomielia (dilatação do canal medular), fraturas e hérnias de disco podem ocorrer nas pessoas com NF1, mas não parecem ser mais frequentes do que na população em geral.
Figura 2 – Alterações na coluna vertebral frequentemente associadas à Neurofibromatose do tipo 1.
Causas
As causas das anormalidades do desenvolvimento ósseo na NF1 ainda estão sendo estudadas, mas algumas possibilidades são:
- Alterações no metabolismo da Vitamina D
- Osteopenia (densidade óssea menor do que o normal)
- Erosão de ossos pela presença dos neurofibromas
- Aumento da pressão dentro da dura máter na medula
- Defeito no desenvolvimento embrionário do folheto mesodérmico por insuficiência da neurofibromina
Ainda não foi possível associar as alterações da coluna vertebral com qualquer tipo de variante genética no gene NF1.
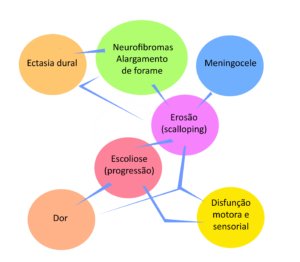
Associação entre as alterações ósseas os sintomas e a progressão da escoliose.
Parece haver uma ligação entre as alterações ósseas, os sintomas e a progressão da escoliose da seguinte maneira: a presença de ectasia dural e de meningocele está associada com neurofibromas nos forames e este conjunto de fatores aumenta a chance de erosões, o que favorece a progressão da escoliose, que causa dor nas costas e redução da função motora e sensorial (Figura 2).
A presença de três ou mais alterações ósseas aumenta significativamente o risco de progressão da escoliose.
Tratamento da escoliose
O tratamento da escoliose requer a avaliação da ortopedia com experiência em NF1 e informações sobre este tema podem ser encontradas em nossa página (ver aqui).
Dr. Lor