Notícias do Congresso NF – Dia 1
Dra Juliana Souza e Dr. Bruno Cota
Temas da Tarde (26/6)
- Schwannomatoses
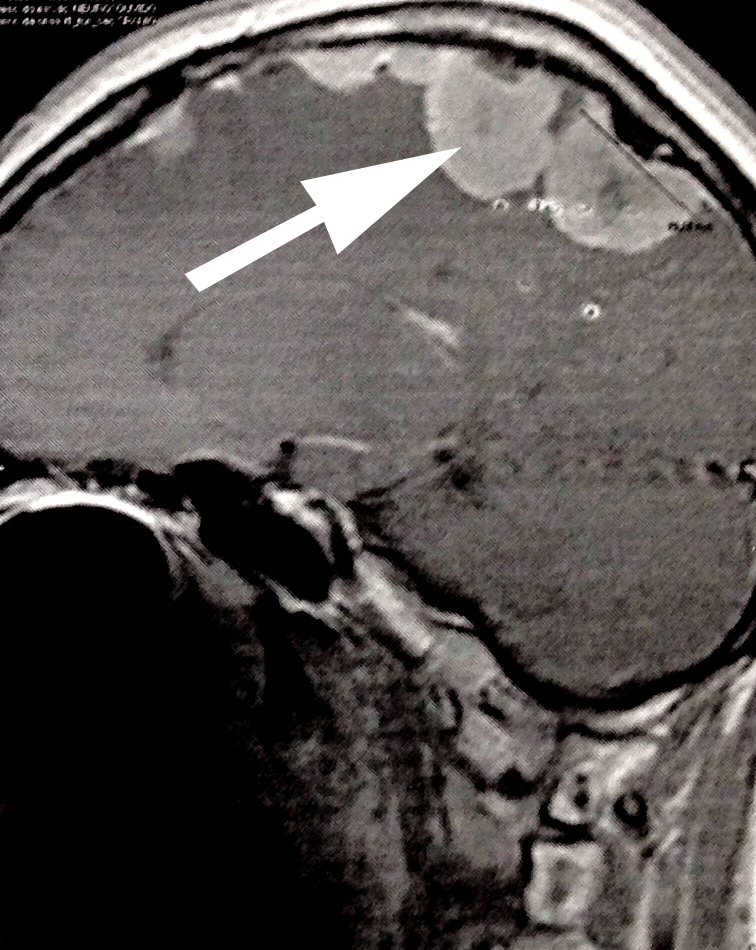
A programação da tarde incluiu uma palestra sobre os meningiomas na schwannomatose relacionada ao gene NF2 (NF2-SWN) – ver abaixo os detalhes da palestra, – uma nova classificação para as variantes patogênicas de significado incerto, o papel das variantes do gene LZTR1 nos schwannomas e em seguida a discussão de casos clínico de schwannomatose e gliomas das vias ópticas em crianças com NF1.
Os aspectos mais relevantes debatidos foram a indicação ou não da radioterapia na NF2-SWN, o uso do Bevacizumabe (ver aqui nossa opinião sobre este medicamento) e a possibilidade do uso compassivo do Brigatinibe em crianças, uma vez que esta droga ainda não foi estudada nessa população e teve melhor resultado com schwannomas não vestibulares e meningiomas (ver aqui nossa opinião sobre este medicamento).
Destacou-se a importância do seguimento clínico, no mínimo anual, dos pacientes com NF2-SWN, com exame físico e audiometria tonal e logoaudiometria. Foi recomendada a ressonância (RM) de crânio a partir dos 10 anos, sendo duas por ano no primeiro ano ou antes, se identificado crescimento rápido de algum tumor intracraniano. Se houver sintomas ou genótipos de alto risco, iniciar RM de coluna a cada 2-3 anos.
Radiocirurgia: ainda controversa para schwannomas na NF2, especialmente pelo risco ainda não bem elucidado de formação de novos tumores e pouco efeito sobre a preservação da audição, embora possa controlar o crescimento do tumor.
Sobre o uso do Bevacizumabe, a discussão também ressaltou o seu uso compassivo, quando não temos outras opções, baseado nos estudos de Plotkin e col. de 2009, com 10 pacientes com NF2-SWN, dos quais seis tiveram alguma redução no tumor, quatro alguma melhora na audição, sendo a metade (somente dois) com esta melhora sustentada após seis meses.
Para o Brigatinibe, como mencionado anteriormente, o estudo INTUITT-NF2 identificou um melhor efeito sobre os schwannomas não vestibulares, com redução da dor, e houve melhora da audição em 35% das pessoas, embora não se saiba se, de fato, melhorou a sua capacidade funcional.
- Caso clínico – Glioma das vias ópticas em criança de 3 anos
Na sessão de gliomas de vias ópticas (GVO) relacionados à NF1, foi apresentado o caso de uma criança com 3 anos, com GVO bilateral, acometendo quiasma e radiação óptica, assintomático. Fundoscopia com palidez do nervo óptico, bilateralmente.
Foi ressaltada a dificuldade em se avaliar clinicamente a visão desse paciente, com os testes padronizados para essa faixa etária, especialmente no contexto de que a criança tinha uma maior dificuldade para cooperar com a realização dos testes devido ao diagnóstico concomitante de autismo.
Foram debatidos o uso da ressonância nuclear volumétrica, considerando os riscos da sedação e contraste, como um parâmetro para se definir a conduta (tratamento quimioterápico ou conduta expectante).
Dentro desse contexto, foi apresentada a baixa precisão da ressonância para se definir tratamento baseado no aumento da captação do contraste pela lesão (45% dos tumores com captação aumentada não se associam a declínio da acuidade visual). Quanto ao volume, por si só, também não é um bom preditor de gravidade clínica em boa parte dos pacientes.
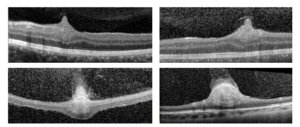
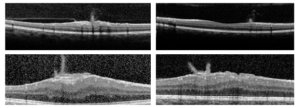
Por outro lado, a OCT pode ser uma boa opção na avaliação de casos como esse, uma vez que a avaliação da camada de fibras nervosas da retina tem uma correlação mais confiável com a qualidade da visão.
Outros pontos debatidos foram a benignidade da evolução da maioria dos casos de glioma das vias ópticas na NF1, diferente da população geral.
Por fim, os estudos recentes com inibidores MEK, mTOR (Everolimus) e Bevacizumabe, como drogas de uso potencial em casos refratários à quimioterapia convencional, uma vez que esta tenha sido bem indicada. Acrescente-se que está em andamento um estudo que visa comparar os resultados da quimioterapia tradicional com o uso de inibidores MEK (iMEK) no glioma óptico.
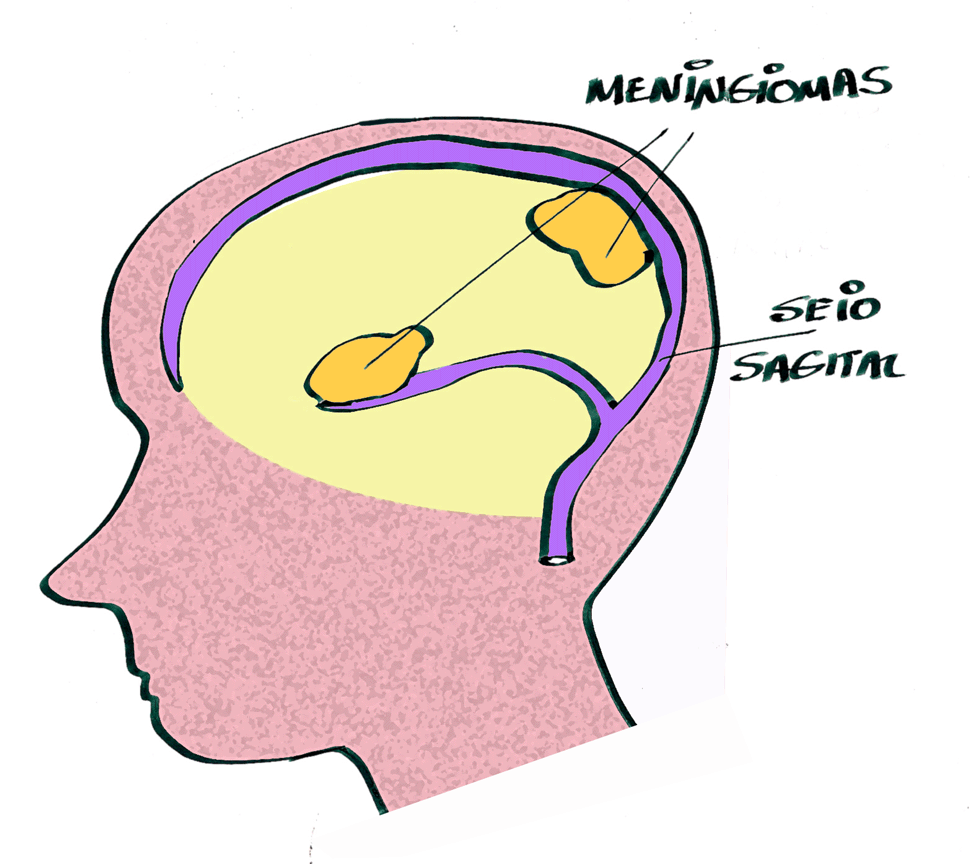
Detalhes da palestra sobre tratamento de meningiomas na NF2-SWN
Apresentada por Brian Na, da Universidade da Califórnia
Introdução com aspectos gerais, como:
- Prevalência (50% dos pacientes com NF2-SWN),
- Risco de 80% ao longo da vida;
- Maioria intracranianos, mas podendo ocorrer na coluna;
- Localização predominantemente supratentorial, sendo os de base do crânio mais comumente observados em crianças.
Acompanhamento dos pacientes
Reforçou a ideia de que o seguimento dos pacientes deve ser individualizado, diante não somente da variabilidade interindividual em relação às manifestações tumorais, especialmente quanto à quantidade de tumores, localização, tamanho e velocidade de progressão.
Embora a maioria dos meningiomas tende a apresentar crescimento lento, foi levantado o questionamento de um possível comportamento um pouco mais agressivo na NF2-SWN, na qual mais da metade dos tumores possuem Grau 2 ou 3 da WHO (ver abaixo nota sobre esta classificação).
Foi reforçado, ainda, a variedade de comportamento dos meningiomas para um mesmo indivíduo, de modo que um ou mais tumores podem apresentar padrões de progressão diferentes.
- Sobre o risco de desenvolvimento dos meningiomas, apresentou o risco de desenvolvimento dos meningiomas baseado na localização da variante patogênica no gene NF2.
- Mostrou que alguns outros genes relacionados ao desenvolvimento dos meningiomas (gene NF2 presente em 50-60% dos meningiomas)
- Sobre o padrão de crescimento, mencionou um estudo de 2023 que distingue os meningiomas em quatro classes baseadas na velocidade de crescimento em 6 meses:
- quiescente (<0,03 cm2);
- linear (>0,03 cm2 em duas medidas subsequentes);
- alternante (quiescente e linear) e
- exponencial.
- Sobre vigilância da progressão, disse que não há um consenso claro sobre a periodicidade das RM, permanecendo a sugestão de individualizar, conforme a velocidade de crescimento, sintomas, e grau WHO (semelhante ao que fazemos no CRNF).
- Sobre tratamento: escassas evidências e pouco consenso para o tratamento além da ressecção dos tumores, apesar de haver 9 ensaios clínicos em andamento, a maioria em fase II, com tratamento imunobiológico direcionado para alguns alvos moleculares (mTOR (3 estudos); RTK (2 estudos); MEK (selumetinibe, 1 estudo); FAK (dois estudos, sendo um deles com Brigatinib) e PI3K/AKT (único de fase II/III).
- Dentre os estudos finalizados, destacou
- Kumthekar, 2022, que investigou o Bevacizumabe para meningiomas recorrentes e refratários (não especificou se ao tratamento cirúrgico ou ao tratamento com radioterapia). Mas não apresentou os resultados claramente desse estudo.
- Outro estudo, com Vistusertib (basket trial para diferentes tumores da SWN NF2, publicado em 2024 – Jordan et al, Neuro-Oncology, com 18 participantes, nos quais a maioria dos meningiomas se manteve estável ou reduziu de tamanho, contudo 12 dos 18 participantes desistiram, por eventos adversos ou “escolha do participante”.
- Mencionou um ensaio clínico de fase III que compara observação pós operatória dos meningiomas grau 2 versus radioterapia (NRG BN-003 e ETORC-1308/ROAM)
- Fez um comentário interessante elucidando que o desfecho na maioria dos estudos tem como principal variável de desfecho a sobrevida em 6 meses (o que não é a principal questão na NF2-SWN, pois não se trata de câncer), ressaltando a importância de ter isso em mente quando se propõe o tratamento clínico vs expectante, considerando a importância de se deixar claro as expectativas realistas para o paciente.
- Propôs considerar a radiocirurgia estereotáxica para alguns casos, citando um estudo que não identificou a ocorrência de neoplasias potencialmente provocadas pela radiação em uma coorte de 267 pacientes com NF2 (mas nesse caso o tratamento direcionado para schwannomas vestibulares, e não para meningiomas); e em análise de outros estudos para tratamento dos meningiomas em progressão destacou em um deles a taxa de controle do tamanho em 5 anos > 90%, mas o controle a longo prazo (>5 anos) variando entre 27% e 51%.
- Ao final, destacou que tais estudos são de baixo valor de evidência devido ao desenho, a maioria deles séries de casos e apresentou um algoritmo baseado no estudo de Wang et al, Neuro-Oncology 2024, para manejo dos meningiomas, baseado em sintomas, risco de compressão e crescimento (discutiremos no CRNF este algoritmo e em breve publicaremos nesta página).
Nota
Classificação da OMS (WHO – World Health Organization) para os meningiomas
Esta classificação é baseada principalmente em critérios histopatológicos e define o grau de agressividade e o risco de recorrência tumoral. A última atualização oficial da OMS é a Classificação de Tumores do SNC de 2021 (WHO CNS 5 edição).
Simplificadamente classifica os meningiomas em:
Grau 1 → Benigno
Grau 2 → Atípico
Grau 3 → Maligno (Anaplásico)
Ver abaixo os detalhes.
Classificação WHO para Meningiomas (2021)
| Grau OMS | Características Principais | Exemplos de Subtipos Histológicos | Prognóstico Geral |
| Grau 1 (Benigno) | Crescimento lento, baixa taxa de recorrência | Meningioma meningotelial, fibroso, transicional, microcístico, secretor, metaplásico, angiomatóide | Excelente |
| Grau 2 (Atypical) | Maior celularidade, mitoses aumentadas (≥4 mitoses por 10 campos de grande aumento), perda da arquitetura típica, necrose focal, maior risco de recorrência | Meningioma atípico, claro, cordóide | Prognóstico intermediário, risco moderado de recorrência |
| Grau 3 (Anaplásico ou Maligno) | Alta atividade mitótica (≥20 mitoses por 10 CGA), atipias marcadas, crescimento invasivo, comportamento agressivo | Meningioma anaplásico, papilar, rabdoide | Prognóstico ruim, alta taxa de recorrência e risco de metástase |
Principais Novidades da Classificação WHO 2021:
- Critérios moleculares começam a ganhar importância, como por exemplo:
- Alterações em NF2, TERT mutações promotoras, CDKN2A/B deleções, etc.
- Essas alterações moleculares podem modificar a classificação prognóstica mesmo dentro de um mesmo grau histológico.
- Subtipos histológicos foram reorganizados, mas os três grandes grupos (Grau 1, 2 e 3) permanecem.
- Classificação baseada também em risco biológico, considerando que nem todo Grau 1 se comporta como benigno a longo prazo (ex: meningiomas localizados em áreas críticas como base de crânio ou com alterações moleculares de alto risco).
AMANHÃ TEMOS MAIS NOTÍCIAS DE WASHINGTON!!!