Como prometi na semana passada, apresentarei a partir de hoje, os resultados clínicos dos estudos sobre o uso de imatinibe no tratamento dos neurofibromas plexiformes.
O mesilato de imatinibe (nome do medicamento genérico) é uma droga bem conhecida pelos oncologistas, que a usam no tratamento de algumas doenças hematológicas (inclusive a leucemia mielógena crônica) e alguns tumores de crianças e adultos.
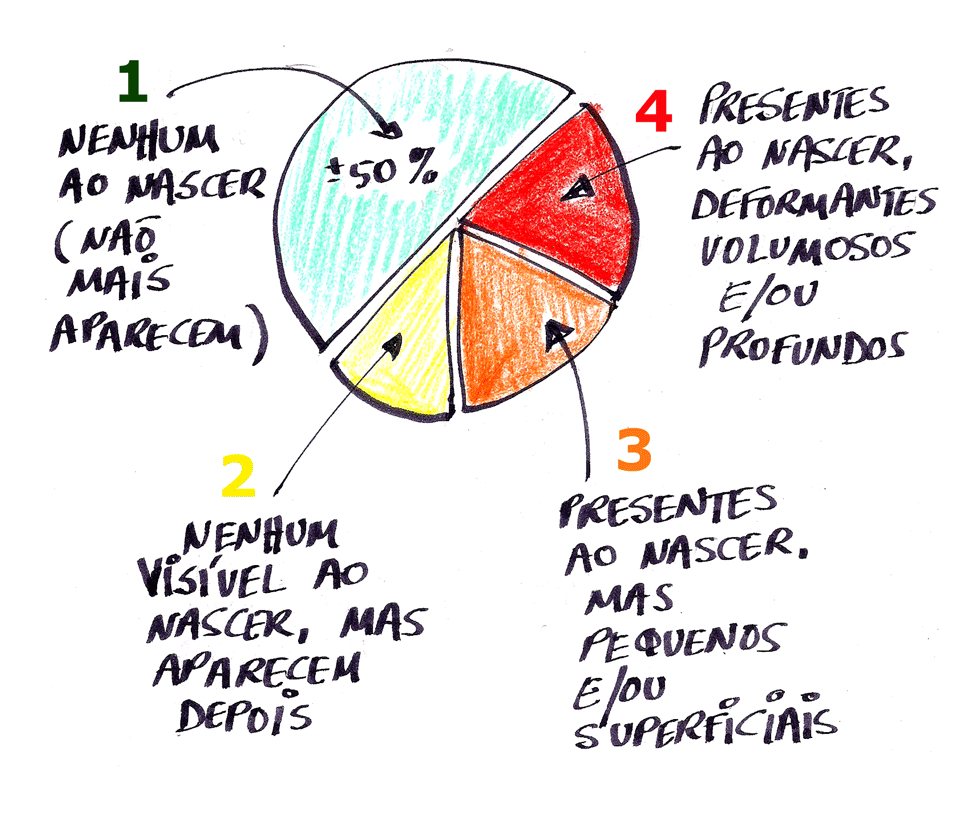
Sabemos que os neurofibromas plexiformes acometem cerca de 50% das pessoas com Neurofibromatose do tipo 1 (NF1), os quais surgem na vida intrauterina em qualquer parte do corpo e são de evolução imprevisível.
Os plexiformes são a principal causa de morte (transformação maligna) e de complicações entre as pessoas com NF1 (dor, deformidades estéticas e perdas funcionais). Tanto para os plexiformes difusos como para os nodulares, o tratamento atual disponível é cirúrgico (ver aqui revisão recente).
A conduta clínica deve ser definida em cada caso em função da localização do plexiforme, dos seus impactos na qualidade de vida da pessoa, da sua taxa de crescimento, dos sintomas que produz e do risco de transformação maligna. Diante de cada uma dessas situações, deve ser pesada a viabilidade da cirurgia e o risco cirúrgico.
O risco cirúrgico é sempre considerável, pois, além das dificuldades técnicas de cada caso (localização, estruturas vitais envolvidas), os plexiformes costumam sangrar muito durante o procedimento cirúrgico e para isto o banco de sangue deve estar de sobreaviso especial.
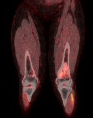
Assim, temos sugerido uma conduta para os cinco níveis de estado clínico dos plexiformes: (1) estável, (2) crescendo sem sintomas, (3) crescendo com sintomas (dor, disfunção neurológica, impacto estético), (4) crescendo com sintomas e risco de morte (sinais sugestivos de grande atividade celular, por exemplo, captação aumentada de glicose no PET CT) e (5) transformação maligna evidente.
Do ponto de vista da cirurgia, podemos considerar quatro níveis de dificuldades técnicas: (A) baixo (tumores superficiais, relativamente bem delimitados, de fácil acesso), (B) médio (tumores internos, porém ressecáveis, sem envolver estruturas vitais), (C) alto (tumores profundos, envolvendo estruturas internas, especialmente torácicas, plexos nervosos e sistema vascular), e (D) inviável (tumores inoperáveis, geralmente de grande volume, difusos e envolvendo estruturas vitais).
(Ver aqui postagem anterior neste blog com a Tabela de sugestão de condutas nos neurofibromas plexiformes nas pessoas com Neurofibromatose do Tipo 1).
Os plexiformes sintomáticos (dor, deformidade e disfunção) e inoperáveis constituem o maior problema para as pessoas com NF1, motivo pelo qual têm sido buscados tratamentos medicamentosos capazes de reduzir a dor, o volume e os seus impactos sobre a saúde.
Com a descoberta de que o crescimento dos plexiformes depende (pelo menos em parte) de um tipo de receptor (chamado de KIT) nos mastócitos, a inibição destes receptores com mesilato de imatinibe foi experimentada em camundongos e diminuiu os plexiformes daqueles animais geneticamente modificados para NF1.
Animados com os resultados obtidos em camundongos, em 2008, um grupo de pesquisadores do Departamento de Pediatria da Universidade de Indianápolis, nos Estados Unidos, tratou com imatinibe uma criança com NF1 em estado crítico por causa de um grande plexiforme na sua cabeça e pescoço, e ela melhorou muito.