


Diante dos cortes de recursos financeiros para a saúde decretados pelo governo Temer, apresento abaixo o discurso que proferi na Câmara dos Deputados em 26 de fevereiro de 2014, durante evento sobre Doenças Raras.


Comentei em outras oportunidades algumas novidades do Congresso de NF de 2017 sobre o problema da dor nas NF (ver AQUI). Hoje trago mais um estudo sobre dor e NF1 que foi apresentado naquele evento pela Dra. Taryn M. Allen e colaboradores do Instituto Nacional do Câncer dos Estados Unidos.
Adaptei abaixo o texto do resumo apresentado. Leia mais

“Dr. LOR, hoje faço uma pergunta sobre o tema do SELUMETINIBE. Sei que não é possível precisar data. Sabemos que as pesquisas demoram e tem etapas a serem cumpridas. Em medicamento pelo que tenho visto acredito que agora entrará na Fase 2 e depois na Fase 3, para depois ser liberada para produção caso seja aprovado. Quanto tempo deve durar esta Fase 2? E quanto tempo deverá estar disponível caso se mostre eficaz? E depois de quanto tempo liberado nos EUA costuma ser liberado no Brasil? Tem como o senhor fazer uma abordagem sobre este tema? E particularmente, se meu filho tiver a oportunidade de entrar em uma das próximas fases da pesquisa, seria melhor na Fase 2 ou na Fase 3? Ou melhor (esperar) chegar ao comércio? ” FP, de local não identificado.
Caro FP, obrigado pela sua pergunta, que considero muito importante. Para responder, preciso recuperar alguns conhecimentos que já temos discutido neste blog.
Este post ficará um pouco mais longo e por isso dividirei em partes que podem ser lidas aos poucos.
Este post também está disponível em inglês, adaptado pelo Dr. Nikolas Mata-Machado da NF Clinic at Amita Health/St. Alexius, em Chicago, Estados Unidos, no site da associação de apoio às pessoas com NF a Neurofibromatosis Midwest (ver aqui AQUI).
Parte 1 – Diferenças entre os neurofibromas
Para começar, lembro que o estudo com o SELUMETINIBE que nos deu esperança (ver aqui) precisa ser repetido por outro grupo de cientistas independentes da indústria farmacêutica que produz o remédio, para que possamos ter mais segurança de que o medicamento traz mais benefícios do que danos às pessoas.
Além disso, os próximos testes com o medicamento precisam levar em consideração se o SELUMETINIBE funciona da mesma forma para os diferentes tipos de neurofibromas plexiformes, se funciona apenas quando os neurofibromas estão crescendo e se funcionam em qualquer idade das pessoas que apresentam neurofibromas plexiformes.
Isto porque o comportamento dos neurofibromas plexiformes varia muito nas pessoas com neurofibromatose do tipo 1 quanto ao tipo, a taxa de crescimento e a idade das pessoas, como mostrou um estudo muito bem realizado em 2012, na Alemanha, pela equipe do Dr. Victor Mautner (ver artigo completo em inglês AQUI).
Os plexiformes podem ser difusos, ou nodulares, ou mistos e uma mesma pessoa pode apresentar um ou mais de cada um destes tipos. Os diferentes tipos apresentam crescimento e complicações variadas e uma mesma pessoa pode apresentar um plexiforme que está crescendo e outro que permanece do mesmo tamanho (ou até se reduz, veja adiante).
No entanto, no estudo em questão, por meio da ressonância magnética tridimensional de corpo inteiro, a equipe do Dr. Mautner examinou todos os plexiformes em conjunto, sem separar em difusos ou nodulares ou mistos.
Parte 2 – Resultados dos estudos na Alemanha
Segundo o estudo do grupo do Dr. Mautner, cerca da metade das 201 pessoas com NF1 apresentou neurofibromas plexiformes, sendo 40% internos, ou seja, visíveis nos estudos de imagem, e 30% superficiais, ou seja, visíveis no exame clínico.
Os plexiformes geralmente são congênitos, ou seja, já estão presentes no momento do nascimento. No estudo, aquelas pessoas que não possuíam plexiformes no início do estudo, não apresentaram novos tumores. Aquelas outras pessoas que já apresentavam plexiformes, desenvolveram novos tumores ao longo do estudo, numa taxa de 1 novo tumor a cada dois anos.
Este resultado sugere que quem não apresenta um plexiforme até o final da infância, provavelmente nunca desenvolverá este tumor ao longo da vida e esta é uma informação importante para as famílias.
Os pesquisadores também verificaram que os plexiformes podem ser pequenos ou grandes em volume: o tamanho médio encontrado foi de 86 mililitros (ou seja, cerca de quatro colheres de sopa), variando do menor com 5 mililitros ao maior com quase 6 litros.
A taxa de crescimento médio dos plexiformes foi de 3,7% ao ano, e foi influenciada pelo volume do tumor: quanto maior o volume inicial, maior a taxa de crescimento, que variou de -13,4% até + 111% ao ano.
Um achado surpreendente, pelo menos para mim, foi a diminuição de 3,4% do volume do plexiforme por ano em cerca de 35% dos adultos, sem qualquer tipo de medicamento ou cirurgia. Embora os autores tenham levantado a possibilidade de erro de medida, esta redução espontânea (ou erro de medida) precisa ser levada em conta nos estudos que testam medicamentos, como o SELUMETINIBE.
A taxa de crescimento dos plexiformes também variou conforme a idade, sendo maior na infância do que depois dos 18 anos. O mesmo grupo de cientistas já havia observado até 20% de aumento de volume por ano em crianças mais novas (ver AQUI resumo do artigo). Por exemplo, uma criança de 5 anos com um plexiforme com o volume de 200 ml (um copo comum) pode apresentar o tumor com cerca de 240 ml um ano depois.
Em torno dos 25 anos a taxa de aumento dos plexiformes já é bem menor, apenas cerca de 0,5% (meio por cento) ao ano. Ou seja, um adulto com 25 anos com um tumor de 200 ml estaria com um tumor de 201 ml no ano seguinte, ou seja, uma mudança praticamente imperceptível a olho nu.
Parte 3 – Efeitos da cirurgia sobre os plexiformes
Num outro estudo científico publicado no ano seguinte (2013), o mesmo grupo do Dr. Mautner apresentou novas informações sobre o comportamento dos plexiformes após tratamento cirúrgico (ver AQUI).
Estes resultados são fundamentais para compararmos os efeitos da cirurgia com o SELUMETINIBE ou outras drogas.
Os pesquisadores estudaram 52 pessoas com NF1, com a média de idade de 25 anos, que foram submetidas à cirurgia para tratamento de neurofibromas plexiformes por causa de dor (20), ou de deformidade estética (21), ou de déficit neurológico (16) ou por estas causas combinadas.
Os principais resultados da cirurgia foram: resolução completa dos sintomas em 46% das pessoas, resolução parcial em 10% e resultado inalterado em 31% dos casos operados. Os tumores voltaram (ou continuaram) a crescer em 23% das pessoas depois da cirurgia, e a análise mostrou que a cirurgia não interferiu na taxa de crescimento dos plexiformes.
Por outro lado, os efeitos indesejáveis da cirurgia foram complicações agudas, como sangramento, em 10% e dificuldades de cicatrização em 5%. Alguns pacientes (13%) desenvolveram novas queixas depois da cirurgia.
O melhor resultado observado com a cirurgia foi quando os médicos conseguiram remover completamente (a olho nu) os plexiformes e isto aconteceu em 25% dos casos, e estes tumores não voltaram a crescer até o final do acompanhamento (cerca de 3 anos). No entanto, estes tumores que puderam ser completamente ressecados eram os menores e mais superficiais e em pessoas acima dos 18 anos.
Em conclusão, os dois estudos comentados sugerem que as crianças com plexiformes mais volumosos apresentam a maior taxa de crescimento dos tumores e que a cirurgia dos plexiformes apresenta seus piores resultados nos tumores maiores e mais complexos (pescoço e cabeça) justamente nesta população.
Assim, os novos estudos com medicamentos (como o SELUMETINIBE) deveriam focar esta população na qual a cirurgia apresenta seus piores resultados.
Em outras palavras, precisam esclarecer se o medicamento deverá ser usado apenas nas crianças ou também nos adultos. Além disso, deveria ser usado apenas nos tumores que estão crescendo ou em todos eles?
Parte 4 – Então, qual seria a duração das novas fases das pesquisas com o SELUMETINIBE?
Não sou capaz de responder precisamente sobre este medicamento, o SELUMETINIBE, porque ainda não conheço os detalhes do projeto de pesquisa. No entanto, de um modo geral, se a meta dos novos estudos continuar sendo a de diminuir em pelo menos 20% o tamanho do tumor inicial, será preciso um tempo de estudo suficientemente grande e parecido com o estudo original para este efeito ser observado, que foi de um ano e meio do uso do SELUMETINIBE.
Outro problema na determinação da duração de qualquer estudo com medicamentos é que ele pode ser interrompido quando se percebe que já se tem as respostas antes do prazo marcado, seja para o bem (efeitos positivos sobre a saúde) ou para o mal (efeitos tóxicos ou piora da saúde).
Portanto, de um modo geral, eu imagino que a duração de cada uma destas próximas fases da pesquisa com o SELUMETINIBE será em torno de 2 anos, para que ele seja definido como uma droga adequada ou não para o tratamento dos plexiformes. Isto, no plano das pesquisas científicas. Se o medicamento se mostrar efetivo, deverá ser submetido aos órgãos de vigilância sanitária nos Estados Unidos e no Brasil, o que pode levar mais um tempo que não sou capaz de definir com segurança.
Finalmente, o leitor FP havia perguntado se seu filho teria a oportunidade de entrar em uma das próximas fases da pesquisa ou se seria melhor esperar o medicamento chegar ao comércio.
Minha impressão é que o mais seguro para qualquer pessoa é receber uma medicação que seja adequada ao seu caso e que seja comprovadamente eficaz e somente depois de ter sido aprovada pelos órgãos reguladores, no nosso caso a ANVISA.
Hoje, responderei a duas perguntas diferentes, mas que têm uma resposta em comum.
“Pelo que tenho lido no seu blog, você defende que as manchas café-com-leite, típicas da NF1, ou já nascem com a criança ou aparecem daí a uns meses, aumentando de número, mas chegando até a desaparecer na idade adulta. No entanto, da minha participação em fóruns, etc, ouço vários relatos de pessoas afectadas dizendo que lhe estão a aparecer mais e mais manchas café com leite. O que diz desta situação?” AG, de Portugal.
“Gostaria de saber se a quantidade de manchas e pintas café com leite tem relação com o grau da NF. E gostaria de saber se existe realmente esses graus na síndrome? Meu filho tem 8 meses e diariamente as manchas e pintas aumentam. Já fizemos o mapeamento é realmente ele é portador da síndrome, ele é mutação já que ninguém tem a NF. Fiz a Ressonância que não apresentou problemas, mas meu filho está atrasado em relação a sentar, rolar e engatinhar..”. K, de local não identificado.
Prezadas AG e K, obrigado pela participação no blog.
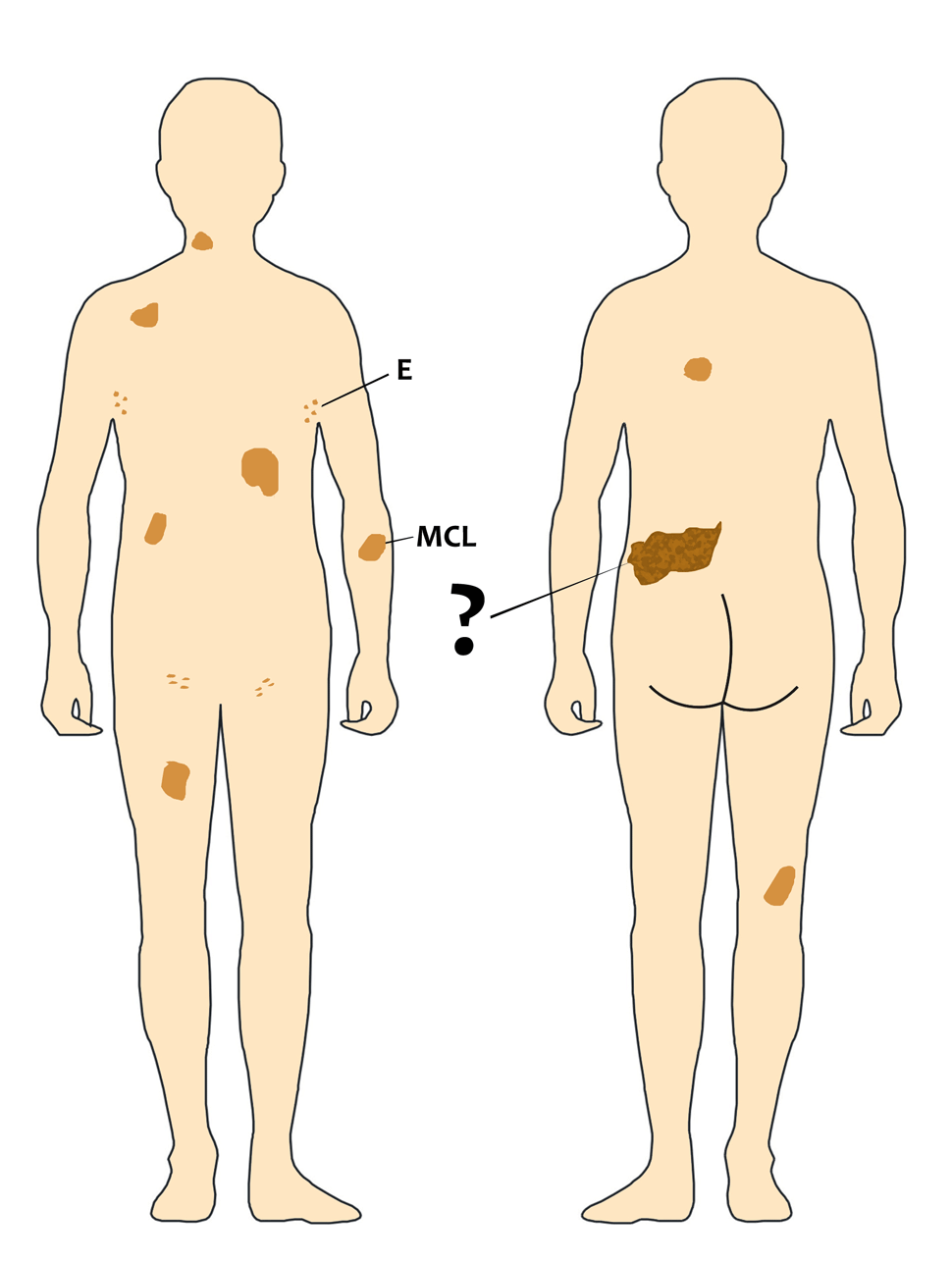
De fato, muitas pessoas relatam o aumento do número de supostas manchas café com leite (MCL) porque geralmente estão se referindo às efélides (E), ou seja, aquelas manchas menores (pintas ou sardas) que estas, sim, na minha experiência, continuam a aumentar ao longo da vida.
Por outro lado, as verdadeiras MCL são congênitas, isto é, estão presentes ao nascimento ou ficam evidentes nas primeiras semanas de vida assim que a criança entra em contado com a luz solar direta ou indiretamente. Minha impressão clínica é de que as MCL não aumentam de número durante a vida e apenas acompanham, proporcionalmente, o crescimento das pessoas. Ao longo da vida adulta e nos idosos as MCL começam a diminuir em pigmentação, ou seja, podem ficar mais claras ao longo dos anos.
Sabemos que a mutação (ou deleção) no gene NF1 é a causa das manchas café com leite e das efélides, mas não sabemos o porquê elas aparecem em certa quantidade e em determinado local. Curiosamente, o aspecto anatômico das MCL é idêntico ao das efélides, fazendo com que alguns estudiosos proponham que sejam considerados um critério único. Do meu ponto de vista, ainda que as MCL e efélides apresentem a mesma histologia, o momento de seu aparecimento (ao longo da infância) e sua evolução (podem continuar a aumentar durante a vida) sugerem que sejam mecanismos genéticos diferentes.
A outra questão é quanto ao significado clínico das MCL e efélides para definirmos a gravidade da doença, o que imagino que seja aquilo que a leitora K. chamou de grau. Até o presente momento, não conheço qualquer evidência científica de que a quantidade e tamanho das MCL e efélides tenham qualquer relação com a gravidade da NF1. Ou seja, a gravidade da NF1 não depende do número, idade de aparecimento e tamanho das MCL e efélides.
Quem desejar saber mais sobre como classificamos a gravidade da NF1 em mínima, leve, moderada ou grave, por favor veja AQUI).
Uma leitora de Portugal pediu minha opinião sobre um estudo que está sendo realizado nos Estados Unidos com uma nova droga para reduzir o tamanho dos neurofibromas plexiformes e sua transformação maligna. O artigo completo em inglês pode ser acessado AQUI .
Agradeço a indicação do artigo, pois ainda não o havia encontrado em minhas buscas por informações científicas sobre NF.
Primeiramente, devo alertar a todos os leitores de que se trata de um estudo ainda e apenas em laboratório, ou seja, a equipe científica está testando a nova droga em linhagem de células humanas com NF1 cultivadas artificialmente. Os resultados são animadores e indicam que o passo seguinte deverá ser testar a droga em camundongos geneticamente modificados para NF1.
Caso os resultados sejam favoráveis nos camundongos, a segurança da droga será testada num grupo pequeno de voluntários humanos. Se for segura, será então experimentada em outras pessoas voluntárias com NF1 para saber se a droga realmente diminui o tamanho dos neurofibromas plexiformes e/ou combate a sua transformação maligna.
Se a droga se mostrar eficiente como tratamento dos neurofibromas neste grupo inicial de voluntários, ela poderá ser oferecida a um grupo maior de pessoas com NF1 e, se os resultados confirmarem sua eficiência, poderá ser submetida às agências de vigilância sanitária nos diversos países.
Depois de aprovada por estas agências a nova droga poderá ser indicada regularmente para as pessoas com NF1 em todo o mundo.
Qual é a ideia da cientista Dra. Kate Barald e seus colaboradores da Universidade de Michigan, nos Estados Unidos?
O grupo considerou que tanto o número quanto o tamanho dos tumores nas pessoas com NF1 aumentam durante a puberdade e a gestação. Como os tumores regridem em tamanho depois da gravidez, a equipe supôs que isto aconteceria em resposta aos hormônios esteroides que estão aumentados naquelas situações.
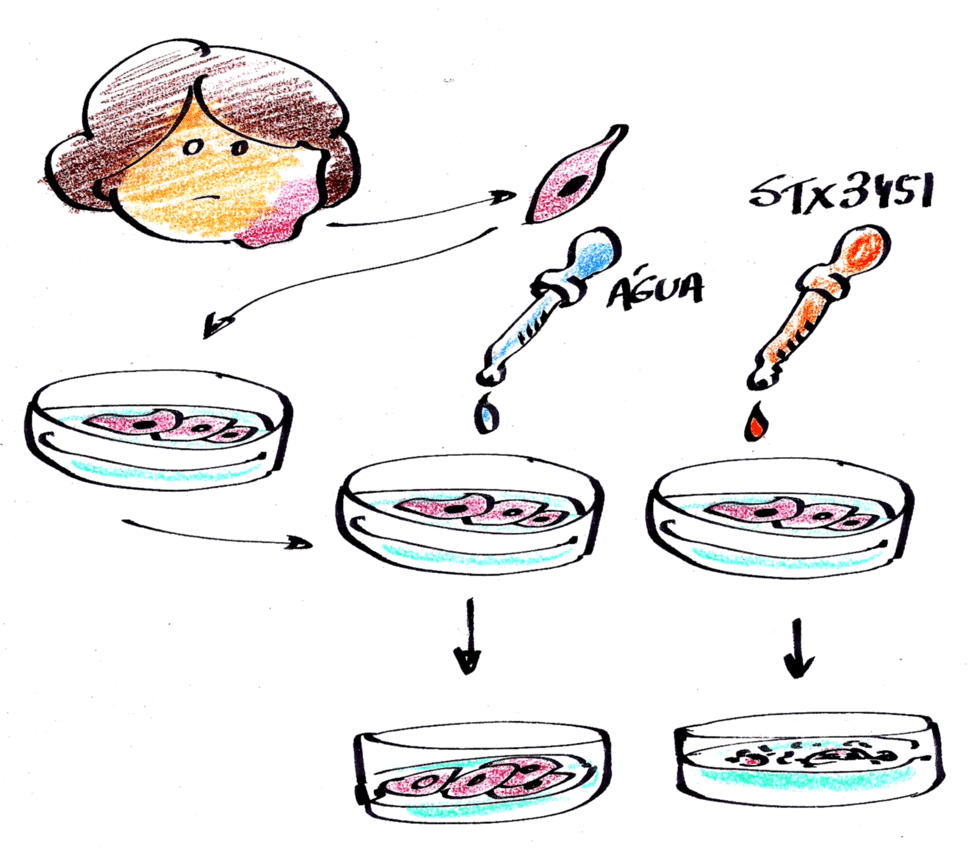
Eles já sabiam que um dos produtos naturais do metabolismo do estrógeno é a substância 2-metoxiestradiol (2ME2) que possui um efeito anticâncer. Então, modificaram esta substância natural e criaram outra chamada STX3451, e testaram seu potencial de ação contra células de tumores recolhidas de pessoas com NF1 e cultivadas em laboratório (ver figura).
Os resultados mostraram que a STX3451 foi capaz de destruir (causar apoptose) tanto as células que foram retiradas de neurofibromas plexiformes (benignos) quanto as células malignas (originárias do tumor maligno da bainha do nervo periférico).
Estes resultados são animadores e merecem nossa comemoração como mais uma esperança de tratamento para as pessoas com NF1.
Vamos torcer para que as novas etapas também sejam positivas.

“Meu filho estava gripado e teve febre alta, levamos ele no Pronto
Atendimento e percebemos que na face onde tem o plexiforme estava sempre mais vermelho e quente. A enfermeira mediu a temperatura com um termômetro de ouvido e sempre apresentava temperatura maior no ouvido direito, que é onde tem o plexiforme, e a diferença era de pelo menos 0,7 graus maior. Ficamos lá por quase 5 horas em atendimento até conseguir baixar a febre, o médico já estava pensando que a febre tinha relação com o plexiforme. Então ficamos em dúvida, é normal o plexiforme durante a febre apresentar temperatura maior? Como vamos saber se a febre tem relação com o plexiforme e o que fazer?” MRP, do Paraná.
Cara M, obrigado pelas informações sobre seu filho.
De fato, o local do neurofibroma plexiforme recebe mais artérias e veias, ou seja, por ali circula mais sangue do que no restante das demais partes mais externas do organismo.
Desta forma, a temperatura no plexiforme se aproxima mais da temperatura interna do corpo (que fica em torno de 37 graus Celsius, sem febre), que é sempre maior do que a da pele (que fica em torno de 32 graus Celsius, na sombra). Assim, se colocarmos a mão sobre a região dos plexiformes mais superficiais, a nossa impressão (ou a medida com termômetro, como a enfermeira fez) é de que a região do plexiforme está mais quente (35 ou 36 graus) do que a pele ao redor.
A temperatura aumentada do plexiforme sem outro sintoma de inflamação (dor, inchação ou mudança da consistência) não é motivo de preocupação.
Ao contrário, um plexiforme que aumenta a temperatura, mas também acelera seu crescimento e apresenta dor (especialmente contínua e noturna) e mudança da consistência (tornando-se mais duro e firme), merece nossa preocupação, pois pode estar em curso a uma transformação maligna.
Sabemos que a transformação maligna é raríssima nas crianças, mas pode acontecer em cerca de 1 em cada plexiforme depois da adolescência ao longo de toda a vida. No entanto, esta transformação é mais comum nos plexiformes grandes, volumosos e profundos.
De qualquer forma, apenas a temperatura um pouco maior na pele sobre um neurofibroma plexiforme superficial não deve ser motivo de preocupação.
Até a próxima semana.

Ontem comentei sobre o estudo realizado na Universidade de Indiana nos Estados Unidos, usando o imatinibe em diversas pessoas com NF1 e neurofibromas plexiformes (quem desejar ver o artigo completo em inglês basta clicar AQUI – em inglês).
Outros estudos mais recentes sobre o uso de imatinibe em neurofibromas plexiformes inoperáveis em pessoas com NF1 apontam na mesma direção, ou seja, de que devemos continuar a estudar mais ampla e profundamente esta possibilidade terapêutica.
Algumas reflexões podem ser feitas sobre os resultados do Dr. Robertson e colaboradores, que deram origem ao nosso atual projeto de pesquisa.
A primeira delas é que não sabemos a taxa de crescimento dos plexiformes antes do tratamento com o imatinibe no estudo do grupo do Dr. Robertson. É possível que aqueles tumores com maior taxa de crescimento apresentem resposta melhor ao imatinibe do que os outros menos ativos? Será que a utilização inicial da tomografia computadorizada com emissão de pósitrons (PET CT) poderia definir o nível metabólico dos plexiformes e indicar os mais adequados para o uso do imatinibe?
Segundo, os autores relatam que o tamanho mínimo dos neurofibromas plexiformes deveria ser de 10 mm, ou seja, tumores muito pequenos para causar grandes riscos, de um modo geral. Não seria mais indicado o imatinibe para pessoas com plexiformes maiores (e, de preferência mais ativos, como vimos acima) e inoperáveis?
Terceiro, não sabemos se houve uma distinção segura entre os neurofibromas difusos (epineurais) e os neurofibromas nodulares (perineurais), os quais possuem origens embriológicas distintas, diferentes momentos de crescimento, diferentes padrões vasculares e possíveis diferenças nas barreiras teciduais à perfusão do medicamento. Não teria sido prudente a separação prévia ou retrospectiva dos efeitos do imatinibe sobre os neurofibromas difusos e os nodulares?
Quarto, sentimos falta de informação sobre os efeitos do imatinibe sobre os sintomas (como dor e disfunção neurológica) e a qualidade de vida das pessoas com o tratamento experimental. Não teria sido mais útil, para todos nós que trabalhamos na clínica e para as pessoas com NF1, se soubéssemos como o tratamento com o imatinibe foi percebido pelas pessoas que o usaram?
Finalmente, quando se busca corretamente a objetividade (medindo-se apenas o tamanho do tumor) não se corre o risco de perdermos informações que talvez sejam mais importantes para as pessoas com NF1, como dor, outros sintomas e qualidade de vida?
Por exemplo, outra pesquisa, realizada com 3 pessoas com NF1 e plexiformes inoperáveis, mostrou que a intensa dor neuropática (presente em muitos plexiformes, especialmente os nodulares), que é de difícil tratamento, praticamente foi eliminada com o uso de outro medicamento (sirolimus), sem que houvesse redução apreciável do tamanho dos tumores (ver aqui o trabalho completo). Portanto, o tamanho do tumor não é o único problema a ser resolvido.
De qualquer forma, a pesquisa do Dr. Robertson e colaboradores (2012) constitui uma base segura sobre a qual podemos formular a proposta de um novo estudo multicêntrico no Brasil.
Pretendemos realizar um estudo com 50 pessoas com NF1 (25 crianças e 25 adultos), as quais utilizariam o imatinibe em neurofibromas plexiformes inoperáveis e sintomáticos e em crescimento (avaliado pelo PET CT), medindo-se o tamanho do tumor e os efeitos clínicos sobre as pessoas, além de levarmos em conta as diferenças entre plexiformes difusos e plexiformes nodulares.
Estamos dando os passos necessários para este projeto, como submetê-lo aos Comitês de Ética em Pesquisa e buscar o financiamento (cerca de 400 mil reais) junto à Universidade Federal de Minas Gerais, à FAPEMIG e ao CNPq.
Quem sabe, algum leitor deste blog tem recursos financeiros para patrocinar este projeto?

Continuando os comentários sobre o medicamento imatinibe, ontem eu disse que um grupo de pesquisadores, liderados pelo Departamento de Pediatria da Faculdade de Medicina de Indiana, em Indianápolis nos Estados Unidos, observou redução dos neurofibromas plexiformes provocados em camundongos geneticamente modificados.
Em 2008, eles resolveram experimentar o medicamento em uma criança com neurofibromatose do tipo 1 (NF1), que estava em estado crítico por causa de um neurofibroma plexiforme na cabeça e pescoço.
Era uma menina de 3 anos de idade, que havia nascido com um grande plexiforme que ocupava sua face, envolvendo a boca e a língua, e se espalhava para trás da cabeça e alcançava o crânio.
O plexiforme envolvia e comprimia algumas estruturas vitais, como a artéria carótida, a veia jugular e vias aéreas superiores. Esta compressão causava sintomas graves de falta de ar, bloqueando o fluxo de ar para os pulmões e interrompendo o sono.
Sei que nem todos entendem imagens radiológicas, mas as figuras abaixo são ressonâncias magnéticas de parte da cabeça e do pescoço, vistas de frente e que foram realizadas antes (1) e depois (2) do tratamento com imatinibe durante 3 meses.
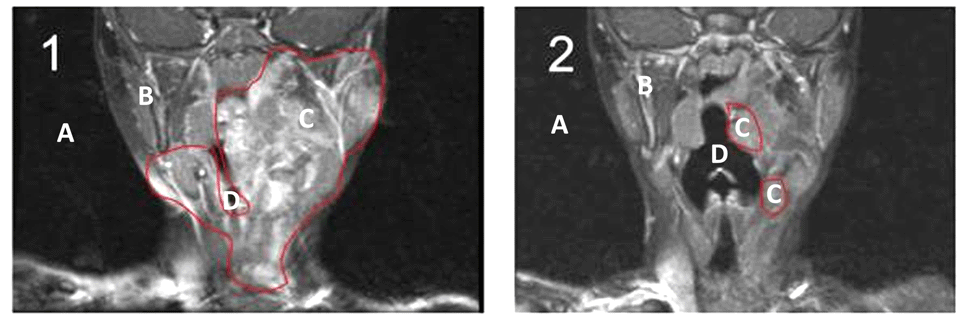
A área mais escura (A) é ar ao redor da cabeça ou dentro das vias aéreas (D), a área cinza é o corpo da menina (B) e a área mais clara (C) circundada por uma linha vermelha é o neurofibroma plexiforme.
Acho que todos podem ver que a área dentro da linha vermelha (C) ficou menor na segunda ressonância (2), realizada depois do tratamento com o imatinibe. É possível observar como o espaço aumentou na via aérea (D), permitindo melhor respiração e sono.
É claro que os pesquisadores ficaram muito animados com este resultado. Quem desejar conhecer o trabalho original, em inglês, publicado numa das melhores revistas científicas do mundo, a Cell, basta clicar aqui.
Os resultados em camundongos e este caso clínico da menina com plexiforme inoperável deram origem a um estudo maior, envolvendo mais pessoas, hospitais e universidades, e que foi realizado sob a coordenação da mesma universidade de Indiana, nos Estados Unidos.
O Dr. Robertson e outros 24 colaboradores conseguiram reunir 36 voluntários, pessoas com NF1 entre 3 e 65 anos de idade, todas elas com neurofibromas plexiformes. As crianças receberam imatinibe por via oral na dose de 220 mg/m2 e os adultos 400 mg/m2 duas vezes por dia durante seis meses.
O imatinibe existe como genérico no Brasil e, apenas como ilustração, do ponto de vista de custo financeiro, hoje, no Brasil, o preço médio do tratamento de uma criança seria de 120 reais por dia e o de um adulto cerca de 250 reais por dia. O medicamento está disponível no SUS para outras doenças, como lembrei ontem.
O objetivo primário do estudo era atingir uma redução de pelo menos 20% do neurofibroma plexiforme, cujo tamanho foi medido em ressonâncias magnéticas repetidas. O estudo foi aprovado eticamente e recebeu financiamento do laboratório farmacêutico Novartis (um dos fabricantes do imatinibe) e da própria Universidade de Indiana.
Eles observaram que seis de todos os 36 voluntários (17%) atingiram o objetivo do tratamento, ou seja, apresentaram redução de pelo menos 20% do tamanho do tumor. Alguns voluntários não conseguiram completar os seis meses de tratamento por diversas razões, mas 23 receberam imatinibe por seis meses, e entre eles seis (26%) também apresentaram 20% de redução de um ou mais plexiformes.
Os efeitos colaterais observados foram: urticária (irritação e inflamação da pele e mucosas) em 5 pessoas, edema e aumento do peso em 6 pessoas, baixa reversível da contagem de leucócitos (neutrófilos) em 2 pessoas, hiperglicemia em uma delas e aumento de uma enzima hepática (aminotransferase) em uma.
A conclusão geral dos autores foi de que o estudo com o imatinibe deve ser ampliado para outras instituições de pesquisa para sabermos se é uma boa opção de tratamento para os neurofibromas plexiformes inoperáveis em pessoas com NF1.
Quem desejar conhecer o trabalho completo, basta clicar aqui.
Amanhã comentarei estes resultados do segundo estudo e o que pretendemos fazer no Brasil.