Apresentei esta situação clínica para colegas com experiência em neurofibromatoses e as respostas estão reproduzidas abaixo na ordem em que chegaram.
Levando em conta os aspectos mais importantes na resposta de cada especialista, ao final resumo algumas condutas que procuram respeitar a maioria das respostas enviadas.
Atenção para a decisão compartilhada
Dra. Luíza de Oliveira Rodrigues (Especialista em Medicina Baseada em Evidências)
A Dra. Luíza lembra a necessidade de adotarmos “a abordagem da Decisão Compartilhada, na qual a opção de investigação genética sempre deve ser discutida. Ela deve ser exposta, com seus prós e contras, para a pessoa e sua família decidirem. Segundo ela, talvez não venhamos a encontrar evidências suficientes para respaldar apenas uma abordagem (aprofundar a investigação) ou outra (acompanhamento clínico apenas) de forma definitiva. Pois o balanço de risco/benefício parece bastante equilibrado. Assim, cabe a nós (equipe médica) somente expor as opções, tentando ajudar a pessoa e sua família a perceberem qual é o seu próprio desejo e/ou necessidade.
Como exemplo, alguns artigos discutem essa questão de uma perspectiva ética. Em um deles, sobre realizar ou não teste genético para diagnosticar uma doença (não NF) que só traria sintomas mais tarde e pra qual só há tratamento quando se manifesta (bastante análogo à situação aqui), observou-se que quando perguntadas, cerca de 1/3 das pessoas queria fazer um teste para sabe o diagnóstico; 1/3 não queria saber; e 1/3 cogitaria fazer o teste. Ou seja, um resultado no qual uma recomendação única não é possível.
Portanto, é fundamental a escuta dos desejos da família e da criança e a exploração dos cenários em relação ao teste e seus resultados”.
Algumas medidas podem ser definidas
Dr. Vincent Riccardi (médico na Califórnia e pioneiro no atendimento das NF)
Dr. Nilton Alves de Rezende (professor da UFMG e fundador do Centro de Referência em Neurofibromatoses do Hospital das Clínicas da Universidade Federal de Minas Gerais – CRNF)
Dr. Riccardi afirma que todas as crianças com menos de 5 manchas café com leite e sem outros critérios para NF1 devem realizar exame oftalmológico para afastar Nódulos de Lisch, alterações da coroide, membranas epirretinianas e opacidades do cristalino.
Ele também recomenda a análise do painel genético para os genes NF1, NF2 e SPRED1, pois o primeiro passo é “identificar uma variante patogênica causal, que nos permite tentar auxiliar as famílias a diferenciar o que são achados da doença, suas manifestações e suas eventuais complicações. Porque as informações que já dispomos nos tornam aptos a reduzir o fardo do diagnóstico para as famílias”.
Dr. Nilton tem uma visão semelhante e concorda com a indicação dos exames oftalmológico e genético, reforçando a necessidade da tomografia de coerência óptica em torno dos 5 anos.
Cuidado com o peso da informação médica
Dr. Bruno Cezar Cota Lage (responsável pelo atendimento no CRNF e doutorando na FM da UFMG)
O Dr. Bruno respondeu que concorda com a Dra. Luíza, embora lembre que há uma tendência dos pais para optarem pelo teste genético.
“Em muitas dessas situações, após eu explicar os prós e contras, a família me pergunta o que eu faria na situação vivida por eles. É uma pergunta difícil de responder, pois tenho que contextualizar o fato de eu ser especialista na doença, como um peso na balança a favor de realizar a investigação genética. Por maior que seja a minha empatia e a compreensão de todo o contexto, não sei se conseguimos a dimensão exata dos sentimentos dos pais. Ao final, nessas ocasiões tenho a sensação um pouco frustrante de que a minha fala final, por mais cuidadosa que seja, interfere diretamente na decisão tomada pelos pais, quando optam por fazer o exame genético.”
A especificidade de cada caso
Dra. Rayana Elias Maia (geneticista, com doutorado em NF)
A Dra. Rayana concorda que é realmente uma tarefa difícil escolhermos uma conduta única, pois é fundamental avaliar caso a caso. Ela lembra que “o tipo, tamanho e distribuição das manchas no corpo vão orientar melhor a conduta. Diante de manchas café ao leite “típicas” sem outro critério para NF1 e nenhuma outra alteração clínica (dismorfias, malformação, baixa estatura/peso ou atraso do desenvolvimento neuropsicomotor) ela tem orientado para que se mantenha uma observação de sinais e sintomas de alerta, programando alguma reavaliação se for uma criança muito pequena.
Tanto na Neurofibromatose do tipo 2 quanto na Schwannomatose (SCW) as manifestações são mais tardias e sem tratamento específico de forma geral. Além disso, na criança sem outros sintomas, considerando que também existe a chance de a investigação vir normal ou com variantes de significado incerto, deve-se discutir com a família a possibilidade de seguimento clínico”.
Dra. Rayana concluiu que, “apesar da angústia para “desvendarmos” o significado das manchas, encontrar variantes duvidosas também gera ansiedade. Além disso, é angustiante chegar a um diagnóstico em uma idade em que não há benefício clínico e que pode acarretar impacto psicológico nos menores portadores e suas famílias diante da incerteza do prognóstico”.
A partir das respostas acima, vejamos a seguir algumas condutas possíveis para crianças com menos de 5 manchas café com leite e sem outros critérios diagnósticos.
1 – Quando as manchas são mais “típicas”
O formato mais comum de manchas café com leite nas pessoas com NF1 são manchas ovaladas, de limites precisos e tonalidade homogênea, geralmente de 10 a 30 mm de diâmetro e algumas podem ter as bordas um pouco irregulares.
- Quando menos de 5 manchas desse tipo estão presentes também em um dos pais (ou ambos, ou irmãos), mas sem quaisquer outros critérios para NF, podemos considerar como diagnóstico provável a mancha café com leite familial (2, 3) (um polimorfismo no gene NF1). Neste caso, sugerimos vida normal.
- No entanto, quando a criança é o primeiro caso na família, precisamos considerar a possibilidade de ser: NF1 em mosaicismo ou Síndrome de Legius em mosaicismo ou NF2 ou Schwannomatose (SCW), pois todas estas situações, EMBORA MUITO MAIS RARAS, podem apresentar manchas café com leite em pequeno número.
- Manchas agrupadas – Se as manchas estão agrupadas em apenas alguma parte do corpo, há uma possibilidade de ser qualquer uma das doenças acima na sua forma em mosaicismo. A foto abaixo foi feita em uma criança com suspeita de NF1 ou Legius em mosaicismo – notar que ela apresenta manchas café com leite e efélides em apenas um segmento do corpo: a parte direita do tórax).
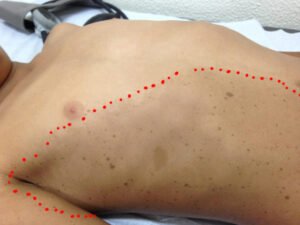
- Manchas espalhadas pelo corpo – Se não há indícios de mosaicismo, há a possibilidade de ser NF do tipo 2 ou Schwannomatose (SCW).
Em todas as situações acima, é preciso considerar na Decisão Compartilhada se devemos dar continuidade à investigação genética ou mantermos apenas a observação clínica por uns anos, monitorando outros sinais ou sintomas.
Devemos lembrar aos pais que NF1 em mosaicismo, Legius em mosaicismo, NF2 e SCW, são doenças ainda incuráveis. Se pedirmos a análise do painel genético e o resultado vier negativo, todos ficaríamos tranquilos. Mas se o resultado for positivo para NF1, Legius, NF2 ou SCW, ele adicionaria um sofrimento antecipado à família, que passaria muitos anos à espera das possíveis e variáveis manifestações destas doenças.
Assim, é preciso discutir com a família se haveria alguma vantagem para a criança em saberem algum destes diagnósticos com tanta antecedência.
2 – Quando as manchas são “atípicas”
Denominamos de manchas café com leite atípicas aquelas que são irregulares, de limites imprecisos e tonalidade variável, muitas delas maiores do que 3 cm.
Estas manchas podem estar ou não associadas a outros sintomas, especialmente neurológicos e musculoesqueléticos.
Quando se distribuem em grandes áreas formando mosaicismos, devemos considerar a possibilidade de ser mosaicismo pigmentar, uma condição congênita de variação na expressão do genoma das células da pele durante a fase embrionária, que podem ser benignas ou estarem associadas a outras doenças genéticas.
Em 2018, uma revisão extensa sobre mosaicismo pigmentar mostrou que uma parte das pessoas (cerca de 30%) apresenta outras alterações clínicas de gravidade variável, indicando a necessidade de complementação diagnóstica.
Assim, devemos discutir na Decisão Compartilhada a indicação de consulta com geneticista para investigar as doenças possíveis sugeridas pelo artigo de revisão.
Finalmente, não devemos esquecer que as manchas café com leite podem estar presentes em outras condições clínicas, como:
- Mancha café com leite isolada (10% da população)
- Síndrome de deficiência de reparo do DNA
- Síndrome de Noonan
- Piebaldismo
- Síndrome de Bloom
- Anemia de Fanconi
Para saber o diagnóstico diferencial destas doenças, VER NOSSO ARTIGO – EM INGLÊS – 2014.
Conclusão
A presença de menos de 5 manchas café com leite numa criança sem outros sinais ou sintomas geralmente significa um bom prognóstico, mas a conduta a ser seguida constitui um desafio clínico para a equipe médica e uma decisão difícil para as famílias.
Dr Lor
Abril de 2022
Referências
- Revisão sobre mosaicismo pigmentar – https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5839061/pdf/13023_2018_Article_778.pdf
- Manchas café com leite familiares Arnsmeier SL, Riccardi VM, Paller AS. Familial multiple cafe au lait spots. Arch Dermatol 130(11):1425-6, 1994.
- Manchas café com leite familiares https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1051784/