Sabemos que as pessoas com NF1 podem passar mal no calor (ver aqui), por isso fiquei feliz em saber que o Sistema Único de Saúde está se preparando para enfrentar as mudanças no clima, inclusive as ondas de calor que estão se tornando mais graves e letais (ver aqui dados de 2026).
Antes de trabalhar com as neurofibromatoses, fui pesquisador durante vários anos na Universidade Federal de Minas Gerais, onde trabalhei com a adaptação humana ao calor, então pretendo contribuir com o esforço do Sistema Único de Saúde (SUS), no presente Governo Lula, para o enfrentamento dos impactos das ondas de calor sobre a saúde pública brasileira.
Por isso, preparei uma cartilha para profissionais da saúde e para familiares de pessoas com NF1.
Se desejar conhecer o que são as ondas de calor e como elas afetam a sua saúde, basta clicar aqui para ver o texto completo.
Selecionei do texto completo três informações sobre as ondas de calor que podem interessar a você.
1 – O que acontece quando o corpo aumenta muito a temperatura (chamamos de hipertermia) numa onda de calor?
Não confundir com FEBRE, veja aqui.
A figura abaixo mostra sinais e sintomas que podem ser moderados (menos de 40 graus centígrados de temperatura interna) ou graves (mais de 40 graus de temperatura interna).
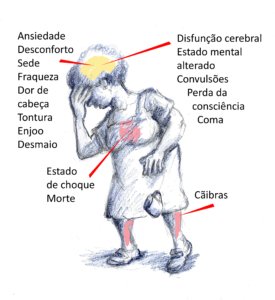
2 – Teste o seu risco
O quadro abaixo mostra um teste simples para conhecer seu risco de passar mal durante uma onda de calor.
| Condição |
Sim |
| Mais de 60 anos ou menos de 10 anos |
|
| Gênero feminino (dobrar pontuação se for gestante ou na menopausa) |
|
| Obesidade (IMC maior que 30) |
|
| Reside em região fria ou temperada |
|
| Sedentarismo |
|
| Mora em casa sem aparelho de ar-condicionado |
|
| Possui doença cardiovascular, neurológica, renal, diabetes, câncer ou NF1 |
|
| Usa algum medicamento para coração, para os rins ou neurológico |
|
| Trabalha em ambiente quente e úmido ou sob o sol |
|
| Já passou mal no calor (desmaio, náusea, confusão mental, “febre”, desidratação, pressão baixa) |
|
| Total |
|
Em termos práticos, quanto maior a pontuação, maior o risco de uma pessoa apresentar hipertermia durante uma onda de calor, portanto, ela deve receber mais cuidados.
3 – E como tratar uma pessoa em estado de hipertermia durante uma onda de calor?
Os procedimentos básicos imediatos para tratamento de uma pessoa com suspeita de hipertermia moderada (temperatura interna menor que 40 graus centígrados) recomendadas pela medicina especializada em ambientes naturais, são:
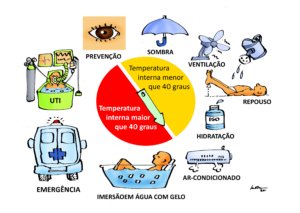
Remoção da pessoa do ambiente quente
Resfriamento natural e rápido com imersão na água fria ou molhando as roupas e promovendo ventilação
Medir a temperatura interna porque ela define se deve ou não ir para o hospital (ver abaixo comentário)
Hidratação oral
Repouso
Provavelmente estas medidas são suficientes para tratamento da hipertermia moderada, mas se houver mais recursos disponíveis, além das medidas acima, recomenda-se:
Remover a pessoa para ambiente com ar-condicionado (menor que 20 graus centígrados)
Resfriar o corpo o mais rápido que puder:
Imersão em água o mais fria possível (quanto mais rápido o resfriamento corporal, menor mortalidade e sequelas)
Se não puder ser feita a imersão, cobrir todo o corpo com compressas de gelo. Se não houver como fazer imersão ou colocar compressas de gelo, usar ventiladores sobre as roupas molhadas (menos eficiente)
Hidratar com líquidos isotônicos ou hipertônicos (para manter o volume sanguíneo e controlar as cãibras) e, se necessário, por via venosa (não há evidência de benefício em se usar infusão gelada)
Elevar as pernas e, se possível, colocar meias de compressão nas pernas para aumentar o retorno do sangue para o coração
Não usar aspirina, nem anti-inflamatórios ou outros antitérmicos!
Nota importante: como medir a temperatura interna?
A medida mais prática da temperatura interna é a medida da temperatura retal. Alternativamente, pode-se usar a temperatura timpânica ou esofagiana. Em último caso, a temperatura axilar pode ser usada, mas sabendo-se que ela pode não refletir exatamente a temperatura interna.
A medida da temperatura interna é um desafio que as equipes de saúde precisam enfrentar daqui em diante, pois há alguns anos, num estudo que fizemos em dois grandes Hospitais de Pronto Socorro em Belo Horizonte, nem mesmo a temperatura axilar era medida rotineiramente no atendimento inicial. O SUS precisa incluir esta norma em suas orientações para enfrentar as ondas de calor.
Na suspeita de hipertermia grave (temperatura interna maior que 40 graus centígrados e/ou outros sintomas graves), usar todos os recursos anteriores enquanto se transfere a pessoa para um hospital de emergências e além das medidas acima, acrescentar:
Suporte ventilatório (com oxigênio)
Suporte cardíaco (com monitorização eletrocardiográfica para detectar arritmias e da pressão arterial)
Medida da temperatura retal (ou timpânica) (maior que 40 graus confirma a gravidade)
Monitorar o volume e a cor da urina para avaliar o estado de hidratação
No hospital deve receber cuidados intensivos porque podem desenvolver as seguintes complicações:
Estado de choque com falência múltipla de órgãos
Encefalopatia (tremores, confusão mental e agressividade durante resfriamento rápido)
Insuficiência respiratória
Lesão hepática e renal agudas
Rabdomiólise (lesão muscular grave)
Coagulação intravascular disseminada
Isquemia intestinal com septicemia
Os detalhes do tratamento intensivo fogem ao objetivo deste texto, mas todas as medidas terapêuticas devem ser tomadas enquanto se continua o resfriamento do paciente. Até a temperatura interna (retal) atingir 38,3 a 38,8 graus centígrados, o resfriamento ativo deve ser mantido (ver detalhes aqui).
As equipes de emergência precisam criar condições técnicas (salas especiais e ambientes adaptados) que permitam o resfriamento por imersão em água gelada enquanto outros procedimentos são realizados.
A equipe médica deve também fazer o diagnóstico diferencial com: hipoglicemia, epilepsia, doença do sistema nervoso central, hiponatremia, hipernatremia, edema cerebral de alta altitude, infecção grave, alteração endócrina grave e ingestão de drogas.
Veja mais informações no texto completo AQUI.
Portanto, cuidemos da vida.
Dr Lor