Este espaço é destinado a opinião de pessoas com experiência em diversos assuntos relacionados com as neurofibromatoses.

Para Luíza, Juliana, Bruno, Luciano…
Uma leitora pergunta por que não estamos realizando novas pesquisas no Centro de Referência em Neurofibromatoses do Hospital das Clínicas da UFMG. Enviei à ela algumas razões que parecem contribuir para que, nos últimos anos, tenha aumentado nossa dificuldade para conseguirmos financiamento público e reduziram as pessoas interessadas em projetos de iniciação científica, mestrado, doutorado e pós-doutorado, apesar de várias perguntas importantes que teríamos para serem estudadas em benefício das pessoas com neurofibromatoses.
Algumas causas dessa redução das nossas pesquisas podem ser imaginadas. Uma delas seria o fato de que nós do CRNF temos nos recusado a entrar na corrida competitiva insana pela publicação de artigos científicos a qualquer preço (ver aqui nossos motivos para isso). Assim, publicando menos do que outras pessoas, pontuamos menos na disputa dos currículos e ficamos no final da lista para financiamentos públicos, apesar de nossas contribuições para o conhecimento internacional no campo das neurofibromatoses (ver aqui).
Outra razão para nossa interrupção nas pesquisas, talvez seja o fato de que também decidimos, há muitos anos, que não realizaríamos nenhuma pesquisa que tivesse financiamento de laboratórios farmacêuticos ou da indústria de equipamentos médicos para que pudéssemos manter nossa autonomia científica para realizar perguntas e obter respostas sem a influência dos interesses econômicos. Assim, estamos na contra mão de outros grupos de médicos e médicas que estão recebendo financiamentos de laboratórios para realizar pesquisas relacionadas com alguns medicamentos chamados inibidores MEK.
Por outro lado, não podemos esquecer que os governos dos últimos anos reduziram o seu investimento em pesquisa e conhecimento, pois para o neoliberalismo a construção do conhecimento e a e a educação pública não são considerados investimentos, mas custos. Com isso, sofremos cortes profundos na ciência e educação na chamada austeridade e responsabilidade fiscal impostas a todos os países (ver aqui Clara Mattei). Por isso, houve redução de recursos nos governos Temer e Bolsonaro e, mesmo no governo Lula, as universidades entraram em greve por melhores condições de trabalho.
No meio destas dificuldades, ainda conseguimos terminar alguns estudos científicos com apoio financeiro da AMANF, aqueles que haviam sido iniciados antes da pandemia, a qual também contribuiu para a quebra da nossa capacidade de realizar pesquisas.
No entanto, outra causa, talvez mais importante, foi publicada em dois artigos (ver aqui e aqui) na revista Nature, que mostraram que, em 10 anos, metade das pessoas que trabalham com pesquisa científica abandonam essa atividade e a maior parte destes abandonos ocorre entre as mulheres cientistas. O motivo? Exaustão geral e machismo no reconhecimento das mulheres.
Este achado indica que estamos diante de um problema mundial: a exploração do trabalho humano, inclusive intelectual, em busca de mais lucro, que aumentou enormemente nos últimos anos, especialmente em função da onipresença da internet, que trouxe o trabalho para casa, para nossas noites e para os finais de semana, fazendo com que a maioria das pessoas tenha passado a trabalhar permanentemente para manter o mesmo salário (ou resistir à sua queda).
Neste contexto de competição e exploração, sufocados pela necessidade de trabalhar mais e mais, não temos tempo para nos dedicar à pesquisa de longo prazo, ao estudo sem resultados tecnológicos imediatos. Perdemos aquele tempo tão necessário para o divagar criativo, para o considerar possibilidades e perguntas novas, para o tentar imaginar respostas para perguntas que todos nos fazem a respeito do conhecimento científico nas neurofibromatoses.
O resultado disso tudo é que, esgotados, renunciamos à busca pelo conhecimento científico para tentarmos sobreviver no trabalho mecânico cotidiano. Exaustos, não temos sequer energia para imaginar um novo modo de fazer ciência, uma nova sociedade, nem um futuro promissor dentro das políticas atuais públicas de investimento no conhecimento e na educação.
Quem desejar compreender melhor este momento, sugiro a leitura do livro “Nas ruínas do Neoliberalismo” da professora norte-americana de ciência política Wendy Brown.
Não estamos sozinhos neste naufrágio da ciência dedicada ao bem coletivo.
Dr. Lor


Dando continuidade à divulgação online dos capítulos escritos para a Edição Comemorativa dos 20 anos do CRNF, a ser lançada em novembro de 2024, apresentamos o esclarecedor texto da Professora Dra. Eugênia Ribeiro Valadares, geneticista que tem nos socorrido frequentemente no CRNF sobre temas relacionados à NF1 e às Schwannomatoses. Agradecemos mais esta colaboração fundamental para a ampliação dos nossos conhecimentos.
Dr. Lor
Eugênia Ribeiro Valadares
Médica pediatra e geneticista, MD, PhD
Professora titular do Departamento de Propedêutica Complementar da Faculdade de Medicina da UFMG (aposentada)
Introdução
Na revisão cronológica das neurofibromatoses feita por Antonio, Goloni-Bertollo e Trídico em 2013, vemos que as manifestações clínicas da neurofibromatose tipo 1 (NF1) foram descritas em 1785, por Mark Akensidi, com relato de um homem com nódulos cutâneos, manchas de pele e cabeça grande. Quase um século depois, em 1882, Friederich Daniel Von Recklinghausen reconheceu a NF como uma entidade nosológica ao descrever dois casos e a doença ganhou seu nome. Já se sabia que a doença tinha o padrão de herança autossômico dominante, mas apenas no final da década de 1980 e início dos anos 90, o gene NF1, responsável pela NF1, foi mapeado no braço longo do cromossomo 17, em 17q11.2.1
Quem desejar conhecer um pouco mais de genética para compreender melhor o texto da Dra. Eugênia Ribeiro Valadares, veja este vídeo O que é o DNA? (youtube.com)
A NF1 é herdada de forma autossômica dominante, embora aproximadamente metade dos indivíduos afetados sejam casos esporádicos causados por uma mutação nova (ou “de novo”) do gene NF1 da linha germinativa, doravante chamada de variante patogênica. Pensava-se que a penetrância de NF1 fosse completa após a infância, mas testes moleculares documentaram penetrância incompleta de variantes patogênicas do NF1 em um pequeno número de indivíduos. Isto significa que a grande maioria das pessoas com uma alteração patogênica em um dos alelos do gene NF1 apresentarão algumas características da NF1. Já a expressividade do fenótipo é extremamente variável, mesmo em pessoas de uma mesma família.
O gene NF1 codifica uma proteína chamada neurofibromina, que funciona na regulação da via de sinalização RAS, que controla a proliferação celular. Na análise genética dos tumores associados com NF1, as células têm variantes patogênicas em ambos os alelos NF1: a variante patogênica da linha germinativa e uma outra variante patogênica adquirida somaticamente do outro alelo. NF1, portanto, funciona como um gene supressor de tumor.
A figura abaixo mostra as variantes patogênicas do gene NF1 (ou seja, “mutações” que podem causar doenças) podem ocorrer em várias partes do gene.
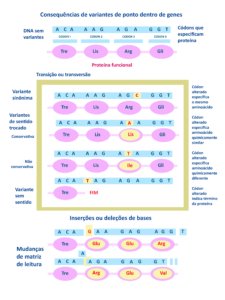
Essas variantes podem estar tanto nas regiões que codificam proteínas quanto em regiões que não codificam. Elas podem incluir diferentes alterações genéticas (ver parte delas na Figura 1):
- DNA sem variantes, proteína funcional, assim a pessoa não apresenta NF1.
- Variantes sinônimas – o códon está alterado, mas especifica o mesmo aminoácido, assim a proteína não sofre alterações.
- Variantes de sentido trocado
- Conservativas – o códon alterado especifica aminoácido que produz proteína quimicamente com a mesma capacidade funcional.
- Não conservativa – o códon alterado especifica aminoácido que produz proteína quimicamente diferente, portanto não funcional – a NF1 se manifesta.
- Variantes sem sentido – o códon alterado indica a parada da síntese da proteína, que assim se torna disfuncional e a NF1 se manifesta.
- Inserções – acréscimos de bases modificam a matriz de leitura impedindo a síntese de proteína funcional – a NF1 se manifesta.
- Deleções – são perdas de partes do gene NF1 e de outros genes próximos.
- Variações no número de cópiasde partes do gene, que podem ser menores e afetar partes internas do gene.
- Alterações nos locais de emenda(locais onde o RNA é cortado e colado), que podem prejudicar a forma como a célula lê o gene.
- Mutações que afetam o início da tradução, ou seja, o ponto onde a célula começa a produzir a proteína a partir do gene.
- Inserções ou deleções complexas, que são mudanças mais complicadas no gene.
- Translocações, que são trocas de pedaços entre diferentes partes do DNA.
- Inserções de elementos móveis, como Aluou LINE, que são pedaços de DNA que podem se mover e se inserir em diferentes partes do genoma.
Em 1988, o National Institute of Health (NIH) estabeleceu critérios clínicos de diagnóstico para a NF1, que precisaram ser modificados em 2000 pelo fato de que 46% das crianças com NF1 não atingiam os critérios diagnósticos com 1 ano de idade, mas 97% delas atingiam esse critério aos 8 anos de idade e 100% delas aos 20 anos. O recurso do diagnóstico molecular para detecção de variante patogênica no gene NF1 pode estabelecer o diagnóstico precoce de crianças suspeitas.
Mais de 100 condições genéticas e síndromes de anomalias congênitas múltiplas que incluem máculas café com leite ou outras características individuais da NF1 foram descritas, mas poucas dessas doenças são confundidas com NF1. A síndrome de Legius, entretanto, pode ser indistinguível da NF1 em uma criança pequena porque os neurofibromas e os nódulos de Lisch geralmente não surgem até mais tarde na infância ou adolescência em pessoas com NF1. O exame dos pais para sinais da síndrome de Legius ou NF1 pode distinguir as duas condições, mas em casos simplex (“de novo”), a reavaliação do indivíduo após a adolescência ou testes moleculares podem ser necessários para estabelecer o diagnóstico.
O mosaicismo na NF1 pode se apresentar como uma doença generalizada ou de forma localizada (segmentar). A NF1 generalizada em mosaico pode ter apresentações semelhantes à NF1 generalizada ou ter um fenótipo mais brando e, portanto, pode ser sub-reconhecida na prática clínica.
Quando solicitar testes genéticos
O teste genético NF1 pode ser realizado para fins de diagnóstico ou para auxiliar no aconselhamento genético e planejamento familiar. No caso de risco de filhos com NF1, o conhecimento da variante patogênica é imprescindível para a fertilização “in vitro” com seleção de embriões.
Se uma criança preenche os critérios de diagnóstico para NF1, a confirmação genética molecular geralmente não é necessária para estabelecer o diagnóstico. Para uma criança pequena que apresenta apenas máculas café com leite, o teste genético NF1 pode confirmar um diagnóstico suspeito antes que um diagnóstico clínico seja possível, ou se for feita a pesquisa em painel, pode detectar variante patogênica no gene SPRED1, da Síndrome de Legius, um dos principais diagnósticos diferenciais com NF1.
Algumas famílias podem desejar estabelecer um diagnóstico definitivo o mais rápido possível e não esperar por novas manifestações clínicas, e o teste genético geralmente pode resolver o problema. Com uma taxa de sensibilidade de 95%, o teste genético é considerado altamente confiável, embora um teste negativo não descarte completamente a condição.
Testes genéticos
Durante muitos anos a doença era diagnosticada apenas por critérios clínicos, pelo fato do sequenciamento do gene NF1 pelo método de Sanger ser dificultado pelo grande tamanho do gene (350 kb, 60 exons), pela alta taxa de novas mutações, pela falta de agrupamento mutacional e pela presença de numerosos loci homólogos. Felizmente o avanço dos recursos genéticos na última década, em especial do sequenciamento de nova geração (next generation sequencing, NGS), facilitou o diagnóstico molecular.
O diagnóstico molecular da NF1 pode ser realizado em qualquer fonte de DNA, geralmente em amostra de sangue, de saliva ou de células da bochecha. Milhares de variantes patogênicas distintas foram identificadas em diferentes pacientes; a maioria leva à perda da função do produto do gene, como esperado para um gene supressor de tumor.
O sequenciamento do gene NF1 pode ser realizado isoladamente, em painéis de exames moleculares que o incluam ou pelo sequenciamento completo do exoma ou do genoma. Geralmente o custo do exame genético do gene NF1 é menor quando realizado em painel de genes. Alguns casos de deleção gênica ou intragênica podem necessitar exame específico para pesquisar deleção, como MLPA ou microarray.
Várias correlações alelo-fenótipo foram observadas em NF1 nos últimos anos:
- A deleção de 1,4 Mb (tipo 1) de todo o gene NF1 está associada a números maiores e aparecimento mais precoce de neurofibromas cutâneos e plexiformes, maior risco de desenvolver tumor maligno da bainha do nervo periférico, anormalidades cognitivas mais frequentes e graves, crescimento excessivo somático e mãos e pés grandes. Um padrão recorrente de características dismórficas que inclui aparência facial grosseira, testa plana, hipertelorismo ocular, ponta nasal larga, orelhas baixas e pescoço largo é frequentemente observado entre adolescentes e adultos.
- Um fenótipo anormalmente grave com neurofibromas plexiformes ou espinhais frequentes, gliomas da via óptica, neoplasias malignas e anormalidades esqueléticas foi observado em adultos com variantes sem sentido de um dos cinco códons entre 844 e 848 [ NM_000267.3 ] que codificam o domínio rico em cisteína-serina da neurofibromina.
- p.Met992del está associado a características pigmentares típicas de NF1, mas não a neurofibromas plexiformes cutâneos, subcutâneos ou de superfície. Um quarto dos indivíduos com essa variante não possui as características clínicas necessárias para atender aos critérios de diagnóstico de NF1, mas tumores cerebrais fora das vias ópticas, deficiências cognitivas ou de aprendizagem e características da síndrome de Noonan ocorrem em frequências semelhantes às observadas em outros indivíduos com NF1.
- p.Arg1038Gly foi associado a características pigmentares leves de NF1, mas a uma escassez de neurofibromas e características frequentes da síndrome NF1-Noonan.
- Variantes sem sentido que afetam p.Met1149 foram associadas a um fenótipo leve caracterizado por características pigmentares, problemas frequentes de aprendizagem e características da síndrome NF1-Noonan.
- Variantes sem sentido que afetam p.Arg1276 foram associadas a uma frequência maior do que o esperado de malformações cardiovasculares, especialmente estenose pulmonar, fenótipo NF1-Noonan e neurofibromas espinhais sintomáticos.
- Variantes sem sentido que afetam p.Lys1423 foram associadas a frequências maiores do que o esperado de neurofibromas plexiformes, bem como de problemas de aprendizagem, malformações cardiovasculares (especialmente estenose pulmonar) e fenótipo NF1-Noonan.
- Várias variantes de sentido errado que afetam p.Arg1809 estão associadas a múltiplas manchas café com leite, mas à ausência de neurofibromas cutâneos ou neurofibromas plexiformes clinicamente aparentes, embora dificuldades de aprendizagem, baixa estatura e estenose pulmonar ocorram frequentemente.
Estudo recente de Garzon e colaboradores (2024) apresenta dados clínicos adicionais sobre 57 indivíduos com a síndrome de microdeleção NF-1, diagnosticados entre 1994 e 2024, durante a infância, adolescência e na idade adulta. Registraram 38/56 (67,9%) com características faciais dismórficas, 25/57 (43,8%) com neurofibromas plexiformes e 3/57 (5,2%) com tumores malignos da bainha dos nervos periféricos no período observado. As manifestações do neurodesenvolvimento mais registadas em indivíduos em idade escolar ou mais velhos incluíram 39/49 (79,6%) com atrasos no desenvolvimento, 35/49 (71,4%) com atrasos na fala expressiva e/ou receptiva, 33/41 (80,5%) com dificuldades de aprendizagem, e 23/42 (54,8%) com déficit de atenção/hiperatividade. Estavam disponíveis dados de testes de QI completos para 22 indivíduos (intervalo: 50-96). Dos 21 adultos desta coorte, 14/21 (66,7%) concluíram o ensino secundário e 4/21 (19,0%) tinham alguma experiência universitária. Os autores concluíram que os resultados cognitivos dos indivíduos com a síndrome de microdeleção NF-1 são potencialmente mais graves do que os descritos anteriormente, mas com incidência similar de TDAH. Além disso, os gliomas da via ótica e tumores malignos da bainha dos nervos periféricos foram diagnosticados numa idade mais jovem na coorte estudada em comparação com a população geral população NF-1, provavelmente como resultado de uma maior monitorização nestes casos.4
Em crianças que apresentam características atípicas, como neurofibromas plexiformes isolados, glioma óptico ou displasia tibial, o teste genético para o gene NF1 também pode ser útil. Nestes casos, o exame genético no sangue geralmente é negativo porque as alterações genéticas podem ter ocorrido apenas no tecido afetado, sendo necessário o teste genético do tecido obtido por biópsia da lesão para identificar a variante patogênica, caracterizando mosaicismo somático.
Crianças que apresentam “NF1 segmentar” também podem ser diagnosticadas com mosaicismo somático por exame molecular do tecido afetado, seja da mancha cutânea ou das células de Schwann de neurofibromas. Este protocolo é útil para adultos que estão preocupados com a possibilidade de transmissão genética por meio do mosaicismo de mutação gonadal NF1 e desejam identificar a variante patogênica específica para futuros testes pré-natais. O diagnóstico molecular de mosaicismo geralmente não orienta o tratamento clínico.
Quadro 1: Resumo das recomendações sobre testes genético
Teste genético para NF1:
- pode confirmar um diagnóstico suspeito antes que um diagnóstico clínico seja possível;
- pode diferenciar NF1 da síndrome de Legius, que é associada ao gene SPRED1;
- pode ser útil em crianças que apresentam características atípicas;
- geralmente não prevê complicações futuras; e
- pode não detectar todos os casos de NF1; um teste genético negativo descarta o diagnóstico de NF1 com 95% (mas não 100%) de sensibilidade.
Aconselhamento genético
Um indivíduo com NF1 herdado de um dos progenitores com variante patogênica na linha germinativa ou por variante patogênica “de novo” tem 50% de chance de ter um filho com NF1 a cada gravidez.
Em contraste, pais não afetados de uma criança com uma variante patogênica “de novo” têm baixo risco de recorrência de ter outro filho com NF1, irmão da criança afetada. Se isto ocorrer, provavelmente trata-se de mosaicismo gonadal para NF1, que é muito raro.
Fertilização in vitro e seleção de embriões
O diagnóstico molecular de pessoa com NF1 torna possível a fertilização in vitro com seleção de embriões sem a variante patogênica.
Referências
- Antonio JR, Goloni-Bertollo EM, Trídico LA. Neurofibromatosis: chronological history and current issues. An Bras Dermatol. 2013;88(3):329-43
- Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR; Council On Genetics; American College Of Medical Genetics And Genomics. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019 May;143(5):e20190660. doi: 10.1542/peds.2019-0660. PMID: 31010905.
- Friedman JM. Neurofibromatosis 1. 1998 Oct 2 [Updated 2022 Apr 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1109/
- Garzon JP, Patete A, Aschbacher-Smith L, Qu’d D, Kelly-Mancuso G, Raski CR, Weisman AG, Hankins M, Sawin M, Kim K, Drackley A, Zeid J, Weaver KN, Hopkin RJ, Saal HM, Charrow J, Schorry E, Listernick R, Simpson BN, Prada CE. Expanding the phenotype of neurofibromatosis type 1 microdeletion syndrome. Am J Med Genet. 2024;e32095.
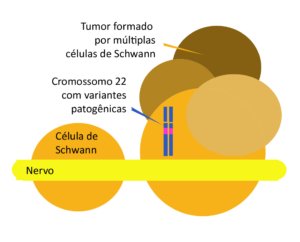
Aspectos genéticos das Schwannomatoses
Durante muitos anos, a NF2 foi confundida com a síndrome mais comum NF1, da qual deriva o seu nome. Nos anos 80, estas duas doenças foram finalmente diferenciadas quando estudos tumorais e análises de ligação localizaram os genes em cromossomas diferentes. O gene NF2 foi clonado em 1993.
Posteriormente outros genes, além do NF2, foram associados a schwannomas vestibulares. A sobreposição dos schwannomas vestibulares que ocorrem na Schwannomatose (SWN) relacionada com o LZTR1 e o mosaico NF2 mimetizando a SWN exigiu uma reavaliação dos critérios de diagnóstico existentes. Depois surgiram o gene SMARCB1 e mais recentemente o gene DGRC8.
Foi criado um comitê internacional para atualização dos critérios de diagnóstico atualizados para as schwannomatoses e atualmente cada doença é classificada de acordo com o gene específico que contém uma variante patogênica.
Por conseguinte, a NF2 passou a ser designada por SWN relacionada com o NF2. Além disso, o comitê recomendou que o termo “Schwannomatose” (SWN) fosse utilizado como um termo genérico para as condições que predispõem ao schwannoma.
Uma vantagem deste formato é que permite a adição de outros tipos de schwannomatoses quando e se novos genes forem identificados.
A recomendação atual é que o nome NF1 permaneça inalterado, e que a NF2 e o outro grupo de doenças relacionadas com a NF fossem renomeados da seguinte: schwannomatose relacionada com ao NF2 (anteriormente designada NF2), schwannomatose relacionada com ao SMACRB1, schwannomatose relacionada com LZTR1, schwannomatose relacionada com 22q, schwannomatose não especificada para doentes que não receberam testes genéticos e schwannomatose não classificada noutra parte para doentes em que os testes genéticos de sangue/saliva e tumores não conseguiram detectar uma variante patogênica.
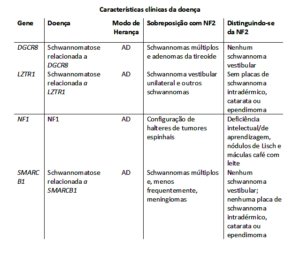
A tabela 1 mostra o diagnóstico diferencial entre as diversas schwannomatoses.
Tabela 1 – Genes de interesse no diagnóstico diferencial da Schwannomatose relacionada à NF2
O gene NF2 e o mosaico somático
A NF2 é causada por alterações inativadoras no gene NF2, localizadas no cromossomo 22q12.2. O gene NF2 de 100 kb é codificado por 17 éxons, tendo sido descritas 10 isoformas resultantes de splicing alternativo em humanos. Além disso, as isoformas alternativas resultam frequentemente de alterações nos éxons C terminal 16 e 17. O crescimento dos schwannomas requer a inativação de ambos os alelos do gene NF2. Os tumores associados à NF2 formam-se quando alterações genéticas somáticas adicionais em células vulneráveis resultam na perda bi-alélica da função NF2 (hipótese de Knudson). No entanto, para promover a tumorigênese, as mutações em NF2 por si só podem ser insuficientes, sendo alterações genéticas adicionais são necessárias. O “segundo golpe” ocorre através da perda de todo o gene NF2 ou da maior parte do cromossoma 22 (2)
As mutações com truncamento da proteína são o evento germinativo mais frequente e causam a doença mais grave. Além disso, a presença de uma proteína truncada está associada a uma idade mais jovem ao diagnóstico e a uma maior prevalência de meningiomas, tumores da coluna vertebral e tumores dos nervos cranianos, exceto do VIII nervo (2).
As alterações patogênicas de NF2 apresentam uma penetrância de quase 100%. Cinquenta por cento dos doentes com NF2 apresentam sintomas e/ou manifestações neoplásicas até os 20 anos de idade, enquanto quase todos os doentes com NF2 apresentam sintomas até aos 60 anos de idade (2).
Suspeita-se que 50% dos casos de NF2 resultem de transmissão hereditária de um progenitor com NF2, enquanto os restantes parecem ser “de novo”, em doentes sem história familiar. Uma mutação pós-zigótica pode resultar em mosaicismo. Os sintomas da NF2 em mosaico são mais leves e frequentemente limitados a uma determinada área ou lado do corpo. As pessoas com mosaico podem apresentar schwannomas vestibulares unilaterais ou doença segmentar. 2
Tabela 1 – Genes de interesse no diagnóstico diferencial da Schwannomatose relacionada à NF2
Testes genéticos
O sequenciamento de NF2 detecta 75% dos casos e a análise de deleção/duplicação direcionada ao gene ou microarray cromossômico detecta 20%. Quando os achados clínicos sugerem o diagnóstico de NF2, as abordagens de testes genéticos moleculares podem incluir testes de gene único ou uso de um painel multigênico que inclua os demais genes relacionados às Schwannomatoses, e análise de microarranjos cromossômicos se necessário (1).
Os testes genéticos para a NF2 não são considerados clinicamente indispensáveis se o diagnóstico puder ser efetuado através de características clínicas. Por outro lado, o teste genético pode ser considerado clinicamente necessário nas seguintes condições:
1) O diagnóstico é clinicamente suspeito devido a sinais e sintomas da doença, mas precisa comprovação.
2) Exame dos familiares em risco sem sinais de doença quando um familiar foi diagnosticado com NF2, especialmente importante para os familiares de primeiro grau de doentes com NF2.
3) Os doentes jovens não cumprem os critérios de diagnóstico sem dados genéticos.
4) Um indivíduo assintomático que fez sequenciamento de exoma ou de genoma para outra indicação é identificado com uma variante patogênica no gene NF2, mas não apresenta características clínicas de NF2. Estes indivíduos e os seus familiares em risco devem ser encaminhados para um centro de referência em Schwannomatose, a variante deve ser determinada como constitucional (linha germinativa), mosaico ou somática, e devem receber aconselhamento genético.
Teste para mosaicismo somático:
Cerca de 25% a 50% dos indivíduos com uma variante patogênica de NF2 de novo têm mosaicismo somático para a variante. Esses indivíduos podem ter testes genéticos moleculares normais de NF2 em tecido não afetado (por exemplo, leucócitos); portanto, testes genéticos moleculares de tecido tumoral podem ser necessários para estabelecer a presença de mosaicismo somático.
Quando o DNA do tumor é testado, variantes patogênicas em ambos os alelos NF2 devem ser identificadas. Isso pode significar testar a perda (ou inativação) de um alelo NF2 avaliando a perda de heterozigosidade. Uma vez que ambas as variantes patogênicas NF2 são identificadas no tumor, o DNA do leucócito pode ser testado para determinar qual das variantes patogênicas é constitucional e qual é somática (ou seja, presente apenas no tumor).
ACONSELHAMENTO GENÉTICO
Os doentes com mutações germinativas do gene NF2 e dos demais associados às Schwannomatoses apresentam uma probabilidade de 50% de transmitir a doença aos seus filhos.
Pessoas com schwannomatoses em mosaico apresentam uma probabilidade reduzida de transmissão aos seus descendentes, devido à esperada ausência de alterações nas células germinativas (espermatozoides ou óvulos).
Fertilização in vitro e seleção de embriões
O diagnóstico molecular de pessoa com schwannomatoses torna possível a fertilização in vitro com seleção de embriões sem a variante patogênica.
Referências:
1) Evans DG. NF2-Related Schwannomatosis. 1998 Oct 14 [Updated 2023 Apr 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1201/
2) Tamura R, Yo M, Toda M. Historical Development of Diagnostic Criteria for NF2-related Schwannomatosis. Neurol Med Chir (Tokyo). 2024 Jun 19. doi: 10.2176/jns-nmc.2024-0067. Epub ahead of print. PMID: 38897938
Continuando a divulgação online dos capítulos escritos para a Edição Comemorativa dos 20 anos do CRNF, a ser lançada em novembro de 2024, apresentamos o a segunda parte do excelente texto da Dra. Vanessa Waisberg, doutora em oftalmologia, que tem sido uma colaboradora incansável do CRNF, atendendo pessoas com NF1 e Schwannomatose relacionada ao gene NF2, que foi justamente o tema de seu doutorado e prêmio nacional de oftalmologia. Agradecemos muito por mais esta colaboração e publicaremos a segunda parte (Problemas Oftalmológicos nas Schwannomatoses) na próxima semana.
Dr. Lor
Dra. Vanessa Waisberg
Oftalmologista com doutorado em
Medicina Molecular pela UFMG
Introdução
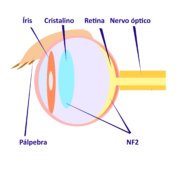
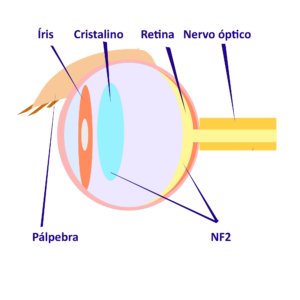
O olho e seus anexos estão frequentemente envolvidos nas Schwannomatoses, entretanto o envolvimento ocular é mais comum na Schwannomatose relacionada ao gene NF2 (SWN-NF2) antes denominada NF2.
Schwannomatose Relacionada ao gene NF2
Um espectro bem definido de manifestações oculares está associado à SWN-NF2. As anormalidades oculares associadas são catarata, hamartomas retinianos, membranas epirretinianas (MER), estrabismo e meningioma da bainha do nervo óptico, como veremos abaixo.
Em crianças com a forma mais grave da doença, as alterações oftalmológicas como estrabismo, catarata, membrana epirretiniana e hamartoma retiniano geralmente estão presentes antes da apresentação neurológica característica e o reconhecimento de tais alterações pode ajudar a reduzir o atraso no diagnóstico dos pacientes com SWN-NF2.
Catarata Juvenil
A catarata aparece em cerca de 60% a 80% dos pacientes e é o principal critério diagnóstico oftalmológico da doença. Tem aspecto característico, com localização subcapsular posterior e/ou cortical. As cataratas geralmente são identificadas na primeira ou segunda década e progridem lentamente ao longo da vida.
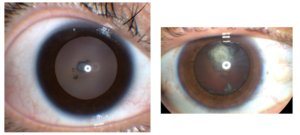
Figura 1 A – Catarata subcapsular posterior em um paciente de 17 anos com SWN-NF2. B – Catarata em um paciente de 50 anos portador de SWN-NF2.
Hamartomas Retinianos
Os hamartomas de retina fazem parte dos critérios diagnósticos da SWN-NF2 desde 2021. Hamartomas de retina são tumores benignos que evoluem lentamente. Geralmente são pequenos, estáveis e não comprometem a visão.
Os hamartomas são identificados no exame de fundo de olho e geralmente aparecem como lesões nodulares e esbranquiçadas, às vezes discretas, e estão associados às formas mais agressivas da doença.
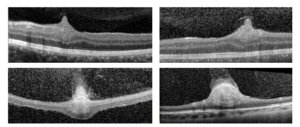
Figura 2. Exemplos de hamartomas de retina em pacientes com SWN-NF2
Membrana Epirretiniana
A membrana epirretiniana (MER) associada a SWN-NF2 tem caraterísticas únicas que a diferencia da MER que ocorrem de forma isolada em pacientes acima de 50 anos. A MER está associada às formas mais graves da NF2 e foi incluída como critério diagnóstico da SWN-NF2 em 2021.
Na SWN-NF2, a MER é espessa e possuiu um aspecto característico em forma de chama de vela que se estende para a cavidade vítrea. Sua localização mais comum é na região central da retina, a mácula, e tende a permanecer estável ao longo da vida, prejudicando pouco a visão, apesar de sua localização central.
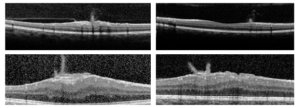
Figura 3. Membrana epirretiniana em “chama de vela”.
Estrabismo
O estrabismo geralmente aparece nos primeiros anos de vida e muitas vezes é a primeira manifestação da SWN-NF2. O estrabismo aparece em cerca de 50% dos pacientes e geralmente está associado as formas graves da doença. O estrabismo pode ser restritivo, quando associado a tumores, ou paralítico, quando associado as paralisias do III, IV ou VI nervo craniano. O tratamento da ambliopia e os resultados cirúrgicos nem sempre são satisfatórios.
Meningioma da Bainha do Nervo Óptico
Os meningiomas da bainha do nervo óptico (MBNO) são tumores benignos que podem ser primários do nervo óptico ou serem secundários a um meningioma próximo ao nervo óptico. Na população geral os MBNO representam cerca de 1% de todos os meningiomas. A acuidade visual tipicamente reduz ao longo dos anos.
A maioria dos pacientes com MBNO associado à SWN-NF2 apresentam redução da mobilidade ocular e baixa acuidade visual precocemente. A acuidade visual geralmente fica reduzida no olho afetado e o tratamento na maioria dos casos só está indicado quando ocorre protrusão significativa do globo ocular.
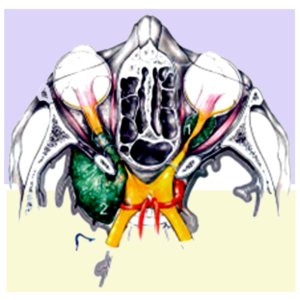
Figura 4. Ilustração evidenciando a diferença do MBNO primário e do meningioma secundário a um meningioma adjacente.
Schwannomatoses não associadas ao geneNF2
O envolvimento ocular nas Schwannomatoses não associadas ao gene NF2 é pouco comum e ocorre pela presença de schwannoma orbitário ou palpebral, sendo os orbitários mais frequentes que os palpebrais.
Nos pacientes que apresentam sintomas como, dor, proptose, diplopia ou redução da acuidade visual, o tratamento é cirúrgico e existe risco de recorrência dos schwannomas orbitários.
As alterações oculares associadas à SWN-NF2, como a catarata juvenil, hamartoma de retina e MER, não são descritas nas outras Schwannomatoses.
Referências:
- Plotkin SR, Messiaen L, Legius E, Pancza P, Avery RA, Blakeley JO, Babovic-Vuksanovic D, Ferner R, Fisher MJ, Friedman JM, Giovannini M, Gutmann DH, Hanemann CO, Kalamarides M, Kehrer-Sawatzki H, Korf BR, Mautner VF, MacCollin M, Papi L, Rauen KA, Riccardi V, Schorry E, Smith MJ, Stemmer-Rachamimov A, Stevenson DA, Ullrich NJ, Viskochil D, Wimmer K, Yohay K; International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC); Huson SM, Wolkenstein P, Evans DG. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022 Sep;24(9):1967-1977.
- Kaye LD, Rothner AD, Beauchamp GR, Meyers SM, Estes ML. Ocular findings associated with neurofibromatosis type II. 1992: 99 (9): 1424-9.
- Bosch MM, Boltshauser E, Harpes P, Landau K. Ophthalmologic findings and a long-term course in patients with neurofibromatosis type 2. Am J Ophthalmol 2006:141 (6): 1068-1077.
- Feucht M, Griffiths B, Niemüller I, Haase W, Richard G, Mautner VF. Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders. Acta Ophthalmol 2008: 86 (8): 882-886.
- Landau K, Yasargil GM. Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol 1993: 77: 646-649.
- Meyers SM, Gutman FA, Kaye LD, Rothner AD. Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc. 1995: 93: 245- 257.
- Feutch M, Kluwe Lan, Mautner V, Richard G. Correlation of nonsense and frameshift mutations with severity of retinal abnormalities in neurofibromatosis 2. Arch Ophthalmol 2008: 126 (10): 1376-1380
- Sisk RA, Berrocal AM, Schefler AC, Dubovy SR, Bauer MS. Epirretinal membranes indicate a severe phenotype of neurofibromatosis type 2. Retina 2010: 30: 51-58.
- Evans, D.G., Mostaccioli, S., Pang, D. et al.ERN GENTURIS clinical practice guidelines for the diagnosis, treatment, management and surveillance of people with schwannomatosis. Eur J Hum Genet 30, 812–817 (2022).