Este espaço é destinado a opinião de pessoas com experiência em diversos assuntos relacionados com as neurofibromatoses.

Somos médicas e médicos no CRNF que acompanham centenas de famílias com Neurofibromatose do Tipo 1 (NF1), uma doença que pode causar neurofibromas. Esses tumores são geralmente benignos, mas podem trazer dor, deformidades e outras complicações. Por isso, estamos sempre em busca de tratamentos que possam controlar ou reduzir esses tumores.
Além disso, em nossa equipe no CRNF, somos 3 médicos e uma médica que têm filhas ou filhos com NF1, portanto, foi com grande interesse que lemos o estudo recente de Chen e colaboradores, publicado na revista científica The Lancet [ver aqui link para o artigo completo em inglês), que avaliou o uso do selumetinibe em adultos com neurofibromas sintomáticos que não podem ser tratados com cirurgia.
Hoje, a cirurgia é o tratamento padrão para alguns casos, mas em tumores muito grandes, espalhados ou em locais difíceis, a cirurgia não é viável. Nesses casos, não há muitos tratamentos disponíveis, o que torna esse tipo de pesquisa especialmente importante.
Uma nova possibilidade de tratamento
O selumetinibe pertence a uma classe de medicamentos chamados inibidores de MEK, que foi a primeira a mostrar alguma redução no tamanho dos neurofibromas. Esse remédio não é uma cura, e tem limitações quanto à eficácia e aos efeitos colaterais, mas representa um avanço. Até agora, ele tinha sido estudado apenas em crianças [ver aqui e aqui], e este estudo traz informações importantes sobre seu uso em adultos.
Resultados principais do estudo
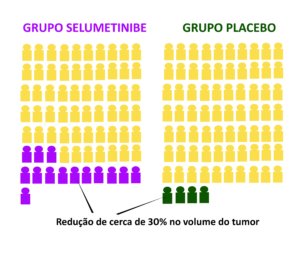
O estudo foi feito em vários centros e envolveu 145 adultos com NF1 e ao menos um neurofibroma plexiforme sintomático e inoperável. Os resultados mostraram que, após 16 ciclos de tratamento, 20% dos pacientes que tomaram selumetinibe tiveram uma redução de pelo menos 34% no volume do tumor. No grupo que tomou placebo (um remédio sem efeito), apenas 5% tiveram alguma redução.
O selumetinibe não foi superior ao placebo na redução da dor, porque houve uma pequena melhora na dor em quem usou o selumetinibe, mas esta redução da dor foi semelhante em quem usou o placebo.
No entanto, o selumetinibe não melhorou a qualidade de vida dos pacientes em comparação com o placebo.
Quais tipos de neurofibromas responderam melhor?
Existem diferentes tipos de neurofibromas: cutâneos (na pele), nodulares e difusos (ver aqui mais detalhes). Esses dois últimos podem formar tumores mais complexos chamados plexiformes, e às vezes afetam todos os nervos que saem da coluna (forma espinhal da NF1).
O estudo não informou se algum desses subtipos respondeu melhor ao tratamento com selumetinibe. Essa informação seria muito útil para orientar melhor os médicos e pacientes sobre quais pessoas têm maior chance de responder melhor ao medicamento.
Por que não foi feito um estudo com troca de grupos?
É uma pena que o estudo não tenha usado um tipo de pesquisa em que TODOS os participantes trocam de tratamento após um tempo (por exemplo, quem começou com placebo passa para o selumetinibe, e vice-versa), o que é comum em estudos com medicamentos de uso crônico.
Só houve uma troca dos pacientes que estavam usando placebo e passaram a usar o selumetinibe.
A falta deste cruzamento limita a utilidade deste estudo da Lancet pois poderíamos ficar sabendo o que acontece quando o tratamento é interrompido: o tumor volta a crescer? O efeito se mantém?
Tolerância ao tratamento em adultos com NF1 de longa duração
Os efeitos colaterais mais comuns do selumetinibe foram problemas de pele, intestinais e no fígado. Cerca de 15% dos pacientes pararam o tratamento por causa desses efeitos. Além disso, o remédio exige monitoramento constante, como exames cardíacos, o que é um desafio, especialmente porque estamos lidando com uma doença crônica e em princípio benigna.
Como os tumores da NF1 geralmente não são malignos, e o remédio só reduz parcialmente o tamanho, é importante pensar com cuidado sobre os riscos e benefícios no longo prazo. Os pacientes devem participar ativamente dessa decisão, e o tratamento deve ser feito com acompanhamento cuidadoso, como ocorre em centros especializados, como o Centro de Referência em Neurofibromatose da UFMG [ver aqui].
Todas as pessoas que precisarem podem ter acesso ao medicamento?
Também lamentamos que o estudo não tenha avaliado se o tratamento é viável do ponto de vista econômico nos adultos. Já sabemos que, para crianças, alguns estudos sugerem que o remédio só seria custo-efetivo se tivesse o preço bastante reduzido ou se o tratamento fosse curto [referências NICE e CADTH]. Mas essa análise ainda falta para os adultos.
Conclusão
O estudo de Chen et al. representa um avanço importante na pesquisa sobre tratamentos para NF1 em adultos. O selumetinibe pode ajudar uma parte dos pacientes com tumores inoperáveis, mas os benefícios ainda são limitados. Há necessidade de mais estudos para:
- Identificar melhor quem tem mais chance de se beneficiar do remédio;
- Entender quais tipos de tumores respondem melhor (nodulares ou difusos);
- Testar combinações de medicamentos diferentes;
- Avaliar os efeitos no longo prazo – o que acontece quando o medicamento é suspenso?
- E verificar se o tratamento é viável do ponto de vista econômico para que todas as pessoas que necessitem dele tenham acesso.
Continuaremos acompanhando os avanços da ciência e informando nossas famílias com transparência e responsabilidade.
Assinam este post a Diretoria da AMANF e a equipe médica do CRNF HC UFMG


SIM, somos a favor da incorporação do selumetinibe pelo SUS, mas com algumas condições, que são importantes para garantir o acesso e a segurança, para quem precisa do remédio, e a sustentabilidade do sistema de saúde:
- Redução do preço em cerca de 90% do preço já listado na CMED (como o Canadá e o Reino Unido conseguiram – ver aqui o link) para que todas as pessoas com NF1 com indicação para seu uso tenham acesso ao medicamento.
- Seja prescrito dentro da bula, ou seja, somente para neurofibromas plexiformes sintomáticos e inoperáveis em crianças de 2 a 18 anos, conforme os pacientes incluídos no estudo original (ver aqui);
- Seja prescrito por profissionais com experiência em neurofibromatoses, sem conflitos de interesse;
- Que seja seguido um protocolo de cuidados que apoie a decisão compartilhada para a decisão do uso do medicamento, que informe claramente aos pacientes os potenciais benefícios e riscos do tratamento (ver aqui os cuidados página da AMANF);
- Que todos os pacientes sejam informados claramente de que ainda não se sabe o que acontece se tivermos que suspender o selumetinibe, inclusive de que há relatos de casos de crescimento acelerado dos tumores com a interrupção do medicamento e isso é um risco a ser considerado (ver aqui relato de caso com o efeito rebote);
- Somente continuar o uso depois de 18 meses de tratamento se houver evidências clínicas objetivas de que o paciente está respondendo ao medicamento, incluindo avaliação objetiva de redução da dor, da melhora da capacidade motora e/ou da redução do tamanho do tumor.
Assinam esta opinião a equipe de médicas e médicos do CRNF e toda a Diretoria da AMANF


Equipe de Apoio Jurídico da AMANF
A importância do trabalho no contexto dos direitos fundamentais está ligada à sua função como meio de realização pessoal, sustento e inclusão social. O trabalho é considerado aspecto essencial para a dignidade humana, garantindo autonomia e participação na sociedade, conforme previsto em instrumentos como a Declaração Universal dos Direitos Humanos e a Constituição Federal, em seus artigos 6º e 7º.
Pessoas com deficiência podem enfrentar barreiras sociais, físicas e atitudinais que limitam suas oportunidades. Por isso, o direito ao trabalho é garantido por uma série de normas legais que visam promover a inclusão e a igualdade de oportunidades no mercado de trabalho, tanto na iniciativa privada quanto no setor público.
Esse é o caso, também, de pessoas com neurofibromatose (NF1 e Schwannomatoses) que, a depender das manifestações da condição, podem ser legalmente reconhecidas como pessoas com deficiência. Esse reconhecimento, sempre realizado de forma individualizada, é essencial para assegurar o acesso aos direitos previstos na legislação brasileira de inclusão.
Acesso a vagas na iniciativa privada
A Lei nº 8.213/1991, conhecida como Lei de Cotas, estabelece que empresas com 100 ou mais funcionários devem reservar uma porcentagem de suas vagas para pessoas com deficiência. Essa reserva varia de 2% a 5%, dependendo do tamanho da empresa, conforme o artigo 93 da referida lei.
Esse dispositivo pode ser invocado por pessoas com neurofibromatose que tenham laudo e avaliação funcional atestando impedimentos duradouros, com impacto na vida profissional.
Acesso a cargos no serviço público
No âmbito do serviço público, a Lei nº 8.112/1990, que dispõe sobre o regime jurídico dos servidores públicos civis da União, das autarquias e das fundações públicas federais, assegura a reserva de cargos para pessoas com deficiência, conforme previsto no artigo 5º, § 2º. Essa norma determina que até 20% das vagas oferecidas em concursos públicos sejam destinadas a candidatos com deficiência, desde que as atribuições do cargo sejam compatíveis com a deficiência declarada.
Além disso, a legislação exige que as provas e avaliações sejam adaptadas, garantindo condições equitativas de participação, como tempo adicional, recursos de acessibilidade ou auxílio de profissionais especializados, conforme a necessidade do candidato. Essa reserva de vagas visa promover a inclusão socioeconômica das pessoas com deficiência, corrigindo desigualdades históricas e garantindo seu direito constitucional ao trabalho digno.
Inclusão no ambiente de trabalho
Além da reserva de vagas, o serviço público e o setor privado devem adotar medidas de acessibilidade e inclusão para garantir que as pessoas com deficiência exerçam suas funções em condições de igualdade. Essas adaptações incluem:
– Acessibilidade arquitetônica: Eliminação de barreiras físicas nos locais de trabalho, como rampas, elevadores, banheiros adaptados e sinalização tátil e visual.
– Tecnologia assistiva: Fornecimento de equipamentos e softwares especializados, como leitores de tela, ampliadores de texto, impressoras em braille e dispositivos de comunicação alternativa.
– Adaptação de materiais e processos: Disponibilização de documentos em formatos acessíveis (braile, audiodescrição, linguagem simples) e ajustes nos métodos de trabalho, quando necessário.
– Flexibilização de horários e jornada: Possibilidade de adaptação na carga horária ou regime de trabalho, conforme as necessidades individuais, sem prejuízo à remuneração.
– Capacitação e sensibilização: Treinamento de gestores e colegas de trabalho para promover um ambiente inclusivo e combater discriminação ou preconceitos.
Essas medidas seguem a Convenção sobre os Direitos das Pessoas com Deficiência (ONU, 2006), internalizada no Brasil com status constitucional pelo Decreto nº 6.949/2009, que determina, em seu artigo 27, a eliminação de barreiras e a garantia de igualdade de oportunidades. Assim, a reserva de vagas vai além da admissão, assegurando condições para permanência no serviço público e na iniciativa privada.
O Estatuto da Pessoa com Deficiência (Lei nº 13.146/2015) complementa essa proteção, proibindo discriminação e exigindo adaptações razoáveis nos ambientes de trabalho, incluindo adequação de espaços físicos, equipamentos e métodos laborais, conforme o artigo 34. A mesma norma prevê a superação de barreiras mediante tecnologias assistivas e ajustes estruturais, conforme o artigo 3º, inciso I.
Conclusão
Em resumo, as pessoas com deficiência têm direito a oportunidades de trabalho em igualdade de condições, com reservas de vagas e adaptações necessárias para superar barreiras físicas, comunicacionais ou atitudinais.
Pessoas com neurofibromatose, quando reconhecidas como PCD, devem ter garantido esse mesmo direito, com pleno acesso às políticas de inclusão laboral. O compromisso com a equidade no trabalho não é apenas uma exigência legal — é um passo necessário para construirmos ambientes mais justos, diversos e humanos, onde cada pessoa possa exercer seu potencial com dignidade.