A escolha pelos melhores tratamentos para a maioria das doenças deve ser feita de forma compartilhada pela pessoa e/ou sua família e os profissionais de saúde, diante do entendimento do contexto, das necessidades e expectativas de cada pessoa e das melhores informações disponíveis a partir dos estudos científicos realizados com um número suficiente de voluntárias e voluntários sofrendo de problemas semelhantes.
Estes estudos científicos são os consensos médicos, que oferecem as orientações sobre os melhores tratamentos. Sabendo que há menos pessoas com doenças raras, menos especialistas e menos estudos científicos direcionados a estas doenças, os consensos médicos são menos robustos cientificamente nas doenças raras.
É o que acontece com as pessoas que sofrem com a Neurofibromatose do tipo 2, a NF2. Atualmente, e a NF2 passou a se chamar Schwannomatose relacionada ao gene NF2 (abreviada como NF2-SWN) e faz parte de um grupo de doenças denominadas de Schwannomatoses – ver aqui as mudanças recentes).
A NF2-SWN acontece por causa de variantes genéticas patogênicas que ocorrem em cerca de 1 em cada 25 a 33 mil pessoas (variando conforme a população estudada). O número de pessoas diagnosticadas com a NF2-SWN é ainda menor: 1 em cada 60 a 70 mil pessoas.
Para exemplificar esta raridade, nos últimos 20 anos atendemos cerca de 70 pessoas com Schwannomatoses entre mais de duas mil e duzentas famílias cadastradas no Centro de Referência em Neurofibromatoses (CRNF) do Hospital das Clínicas da Universidade Federal de Minas Gerais. Ou seja, apenas 2% da população por nós atendida.
Então, a NF2-SWN é uma doença rara e por isso a maioria dos médicos não está bem informada sobre a sua evolução. Sabemos que 95% da experiência dos neurocirurgiões é com o tratamento dos schwannomas esporádicos, ou seja, aqueles que surgem em torno dos 50 anos e apresentam comportamento diferente daqueles encontrados nas pessoas mais jovens com NF2-SWN. Quem desejar mais informações sobre o tratamento dos schwannomas esporádicos ver um consenso internacional aqui – artigo completo em inglês.
Assim, são raros os profissionais da saúde familiarizados com a NF2-SWN, o que gera incerteza sobre o melhor tratamento e por isso as pessoas e suas famílias costumam ouvir opiniões divergentes ao consultar médicos diferentes.
Diante disso, nossas sugestões de tratamentos são orientadas pelos consensos médicos internacionais baseados em experiências clínicas maiores do que a nossa. Por exemplo, o grupo de cientistas que publicou o consenso de 2012, no qual nos baseamos, reuniu dados de mais de 500 pessoas com NF2-SWN (ver aqui o texto original em inglês).
O principal problema na NF2-SWN
Nas pessoas com NF2-SWN surgem tumores nos ramos vestibulares dos nervos vestíbulo cocleares (ver na Figura 1 abaixo a localização destes nervos), chamados schwannomas (daí o nome da doença), que geralmente são bilaterais (85%), crescem lentamente e costumam ser diagnosticados a partir da segunda década de vida.
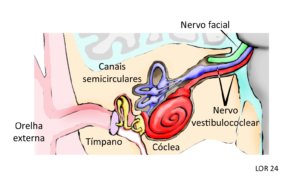
Figura 1 – Localização esquemática do nervo vestibulococlear nos ossos do crânio e sua relação com o cérebro. O ramo vestibular (em lilás) conduz para o sistema nervoso central os sinais neurais gerados nos neurônios dos canais semicirculares pelas mudanças posturais. O ramo coclear (em vermelho) conduz os sinais neurais gerados nos neurônios da cóclea pelos estímulos sonoros que chegam pela orelha externa ao tímpano e são transmitidos pelos ossículos da orelha média. Observa-se o nervo facial surgindo próximo do nervo vestibulococlear e esta proximidade é muito importante na qualidade de vida das pessoas com NF2-SWN.
Os schwannomas vestibulares podem causar perda de audição, zumbido, desequilíbrio, paralisia facial, dor de cabeça e outros problemas neurológicos, inclusive alguns potencialmente fatais.
Nas pessoas com NF2-SWN pode haver schwannomas também em outros pares de nervos cranianos e nas terminações nervosas cutâneas, outros tumores no sistema nervoso chamados meningiomas e ependimomas, além de alterações oculares. Veja em nossa página mais informações sobre o tratamento destes outros problemas.
Em 2020, a Associação de Neuro-Oncologia da Europa criou uma comissão de especialistas que publicou novo consenso sobre a NF2-SWN (ver aqui o artigo completo em inglês). No entanto, nesse consenso, apenas 15,5% dos estudos são especificamente relacionados à NF2-SWN.
O presente texto procura atualizar as orientações que estavam disponíveis em nossa página baseadas no Consenso de 2012, adicionando algumas informações mais recentes deste estudo de 2020.
REGRAS FUNDAMENTAIS
Há duas regras fundamentais no tratamento das pessoas com NF2-SWN:
Regra 1: não se faz cirurgia, nem quimioterapia e nem radioterapia num schwannoma somente porque ele foi encontrado;
Regra 2: não se faz cirurgia, nem quimioterapia e nem radioterapia num schwannoma antes de saber seus efeitos clínicos (surdez, desequilíbrio, zumbido, paralisia facial, compressão do tronco cerebral, hidrocefalia, por exemplo) e sua taxa de crescimento (ou seja, é preciso repetir a audiometria e a ressonância magnética do encéfalo em seis meses para saber a evolução clínica de cada schwannoma).
Por quê?
- Porque as manifestações clínicas são mais importantes para a qualidade de vida das pessoas do que os resultados de exames.
- Porque muitos dos tumores na NF2-SWN podem nunca crescer o bastante para dar sintomas e nunca precisarem ser removidos.
- Porque nem sempre há relação entre o tamanho de um schwannoma e os sintomas (por exemplo, a audição pode ser pior do lado do tumor menor).
Não se sabe ainda ao certo o mecanismo de perda de audição nas pessoas com schwannomas vestibulares. Um estudo científico mostrou que a presença de um schwannoma vestibular esporádico produz alterações anatômicas na cóclea e redução da audição de forma independente do tamanho do tumor. Talvez o mesmo ocorra na NF2-SWN.
- Porque não sabemos qual será a velocidade de crescimento de cada schwannoma.
Em média, o maior crescimento observado é de 2,9 mm por ano e apenas metade dos schwannomas apresentam crescimento num período de 5 anos, enquanto a outra metade permanece estável.
A mesma pessoa pode apresentar crescimento em um dos tumores e nenhum crescimento no outro durante vários anos (ver exemplo abaixo).
- Porque os schwannomas vestibulares não se tornam tumores malignos, ou seja, não dão metástases, não invadem outros tecidos, embora possam comprimir as estruturas ao lado.
No entanto, a exposição à radioterapia ou radio cirurgia pode ser uma causa de transformação maligna.
- Porque as complicações que ameaçam a vida (compressão de tronco cerebral e hidrocefalia, por exemplo) se desenvolvem geralmente de forma lenta.
A compressão do tronco cerebral, por exemplo, praticamente nunca acontece como primeira manifestação da doença.
No entanto, pode haver surdez progressiva em poucas semanas em casos raros.
- Porque a NF2-SWN ainda não tem cura, mas podemos manter a qualidade de vida das pessoas.
Para a maioria das pessoas, é melhor conviver com uma redução da audição do que com uma paralisia facial, que ocorre em cerca de 30% das cirurgias ou com o zumbido pós-operatório, que ocorre em 40% das pessoas.
- Porque existem estudos em busca de medicamentos para os schwannomas, então temos que ganhar tempo sem intervenções que possam deixar sequelas, até que algum tratamento demonstre ser bastante eficiente.
- Porque a maioria dos tratamentos atuais apresenta sequelas permanentes, então qualquer tratamento somente deve ser feito quando houver garantia de benefício.
Ou seja, o resultado do tratamento (por exemplo, melhorar a audição) deve ser mais provável e relevante para a pessoa com NF2-SWN do que a possível sequela do tratamento (por exemplo, a paralisia facial ou o zumbido).
Habitualmente, a cirurgia não recupera a audição já perdida, pois a maioria das pessoas é operada quando os tumores são maiores e a perda auditiva já está presente.
- Porque as cirurgias atuais dependem muito da experiência das equipes cirúrgicas e a maioria delas conhece melhor os schwannomas esporádicos (95% de sua experiência) do que aos schwannomas que surgem nas pessoas com NF2-SWN.
Em geral, as equipes de neurocirurgia pouco familiarizadas com a NF2-SWN ainda chamam os schwannomas vestibulares de “neurinoma do acústico”.
Todas as técnicas cirúrgicas disponíveis atualmente apresentam alta taxa de recorrência dos schwannomas e todos os medicamentos testados até agora ainda apresentam apenas melhora parcial dos sintomas.
Com estas informações em mente, vejamos algumas recomendações para as principais manifestações clínicas dos schwannomas vestibulares nas pessoas com NF2-SWN.
AUDIÇÃO
A redução ou perda da audição é geralmente o sintoma inicial e o mais importante para uma pessoa com NF2-SWN.
A capacidade de compreender a fala humana é a principal função da audição e por isso ela é a medida da qualidade de vida nas pessoas com NF2-SWN.
A perda auditiva geralmente ocorre lentamente, muitas vezes apenas de um lado, e cerca de 4 anos depois de diagnosticada a metade das pessoas com a NF2-SWN apresenta dificuldade importante para compreender a fala humana.
A audiometria é o principal exame complementar para a orientação clínica porque fornece uma medida objetiva da audição.

Figura 2 – Audiometria de uma pessoa com NF2-SWN. Observa-se perda auditiva em ambas as orelhas, mas maior na orelha direita. Perdas maiores do que 10 decibéis (dB) em duas ou mais frequências contíguas ou maiores do que 15 decibéis em qualquer frequência única sugerem o diagnóstico de schwannoma vestibular (com sensibilidade de 93% e especificidade menor que 70%).
A partir do resultado da audiometria, podemos classificar as perdas auditivas de acordo com o Quadro 1, uma classificação utilizada internacionalmente.

O Quadro 1 é apenas uma orientação, porque as pessoas toleram de forma diferente sua perda auditiva. Algumas se sentem muito mal no grau 2, enquanto outras suportam relativamente bem o grau 3.
Por isso, a pessoa com NF2-SWN deve escolher o momento em que a sua perda da discriminação da fala se tornou um problema tão importante que precisa ser corrigido, mesmo com os riscos de algumas sequelas.
A capacidade de discriminação da fala deve ser a informação necessária para ajudarmos a pessoa com NF2-SWN a decidir se deseja ou não receber a intervenção cirúrgica ou medicamentosa.
Para isso, devemos indicar as audiometrias seriadas (a cada seis meses, se não houver novos sintomas antes).
A progressão rápida da perda auditiva (de um grau ou mais em seis meses, por exemplo) é um dos critérios que devem ser levados em conta para a decisão ou não de tratar (ver abaixo um exemplo verdadeiro em nossa experiência).
Esta perda auditiva progressiva e relevante para a pessoa com NF2-SWN combinada com o tamanho do tumor é que orientam nossas sugestões terapêuticas.
Zumbido
Em algumas pessoas o zumbido pode ser o primeiro sintoma, acompanhado ou não da perda auditiva. Dependendo da progressão da perda, nos primeiros momentos o incômodo pode ser leve, a pessoa não perceber e continuar sentindo o zumbido, achando que uma hora vai passar, sem relacionar com uma possível doença.
É importante que o zumbido não seja banalizado, pois ele pode ser um indicativo de perda auditiva. Há pessoas que não desenvolvem o zumbido, mas noutras ele pode ser o sinal mais importante (11%).
TAMANHO DO TUMOR
Sabendo que não existe maneira de conhecer o comportamento futuro de um schwannoma que acaba de ser diagnosticado, o método de escolha para avaliar o tamanho dos schwannomas vestibulares é a ressonância magnética (RNM) seriada a cada 6 ou 12 meses (ou antes, se surgirem novos sintomas neurológicos).
Com o resultado da RNM podemos classificar o tamanho do schwannoma segundo a Figura 2.
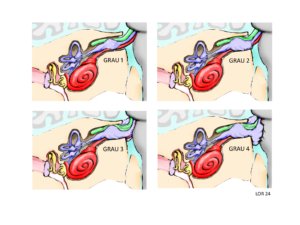
Figura 3 – Escala de Koos para schwannomas vestibulares nas pessoas com NF2-SWN. Grau 1 – Tumor pequeno, restrito ao canal auricular interno. Grau 2 – Tumor médio, avançando para dentro do ângulo bulbopontino, mas sem contato com o tronco cerebral. Grau 3 – Tumor grande, ocupando a cisterna bulbopontina, mas sem contato com o tronco cerebral. Grau 4 – Tumor muito grande com deslocamento de nervos e compressão do tronco cerebral. Observa-se a progressiva compressão do nervo facial com o crescimento do schwannoma.
Esta classificação nos ajuda a compreender algumas situações:
- Os tumores de grau 1 a 3 ainda não ameaçam a vida e por isso podem ser analisados com mais tranquilidade do que os schwannomas maiores (Grau 4).
- Quanto maior o tumor, maior a dificuldade de preservar a audição e o nervo facial, porque o nervo facial está muito próximo ao nervo vestíbulo coclear.
- A localização do tumor é 80% das vezes no ramo vestibular (equilíbrio) e 20% no ramo coclear (audição), apesar da audição estar mais afetada do que o equilíbrio (este dado precisa ser confirmado nas pessoas com NF2-SWN).
- As técnicas cirúrgicas variam de acordo com o tamanho do tumor.
QUAIS SÃO OS TRATAMENTOS POSSÍVEIS?
A ideia principal no tratamento dos problemas causados pela NF2-SWN é que estamos diante de uma doença:
- Crônica (ainda sem cura cirúrgica ou medicamentosa)
- Limitante (especialmente pela surdez e sequelas cirúrgicas, tumores medulares)
- Potencialmente fatal (compressão do tronco, hidrocefalia, cirurgias, convulsões)
- Hereditária (há chance de 30 a 50% de passar a variante patogênica para um filho ou uma filha)
Assim, precisamos pensar nos cuidados médicos no longo prazo, procurando antecipar as possíveis complicações e limitações.
A associação The Children’s Tumor Foundation publicou uma cartilha (ver aqui – em inglês) com orientações importantes para as pessoas com NF2-SWN, que adaptamos para a população brasileira abaixo.
Coordenação clínica
O primeiro passo para o tratamento, depois do diagnóstico confirmado de NF2-SWN (ver aqui), é a escolha de um (a) médica (o) com experiência em neurofibromatoses para coordenar a observação ativa (ver abaixo), as reavaliações, intercorrências, exames e especialistas que podem ser necessários ao longo da vida.
Grupos de ajuda
A participação em grupos de pessoas com neurofibromatoses pode ser fonte de informação e conforto psicológico. Participe da Associação Mineira de Apoio aos Portadores de Neurofibromatoses (AMANF – clique aqui) ou de outras associações semelhantes que existem em vários países.
Aconselhamento genético
É sempre importante realizar o painel genético para identificar o diagnóstico preciso da Schwannomatose (ver aqui).
Sabendo que a NF2-SWN é hereditária (30 a 50% de chance de transmissão da variante patogênica a descendentes), é fundamental ajudar as pessoas no controle de natalidade e planejamento familiar, incluindo a discussão sobre a fertilização in vitro com seleção de embriões.
O diagnóstico da NF2-SWN numa pessoa que já possui filhas ou filhos traz uma questão delicada sobre se deve ou não ser investigada a transmissão da variante para descendentes e quando seria o momento adequado para fazer esta investigação. Geneticistas estão habilitadas (os) a orientar eticamente estas decisões.
A sugestão do Children´s Tumor Foundation é de que crianças com possibilidade de terem herdado uma variante patogênica da NF2-SWN devem realizar:
- Avaliação oftalmológica nas primeiras semanas de vida e anual até os dez anos de idade.
- Ressonância magnética se houver algum problema neurológico e em torno dos 10 aos 14 anos.
- Painel genético depois dos 18 anos ou quando puder tomar esta decisão.
Por enquanto, o SUS ainda não oferece nem o exame genético e nem a fertilização in vitro, o que tem sido uma pauta entre as lutas de associações de pacientes, como da AMANF.
Oftalmologia
A possibilidade de opacificação do cristalino nas pessoas com NF2-SWN (chamada catarata juvenil ou catarata subcapsular posterior) exige que a capacidade visual seja regularmente avaliada por oftalmologista experiente em neurofibromatoses (ver mais informações aqui).
Fonoterapia
A terapia fonoaudiológica pode ajudar na convivência com a baixa audição e com o zumbido. Além disso, pode orientar na aquisição da linguagem labial e na linguagem de sinais, que devem ser aprendidas enquanto há audição.
Além disso, a fonoaudiologia pode ajudar na adaptação de atividades da vida diária, como a vida social, uso de telefones e televisão.
Apoio psicológico
Os impactos psicológicos da NF2-SWN são profundos e a pessoa acometida assim como sua família geralmente necessitam de apoio especializado para conseguir superar as limitações e levarem uma vida com boa qualidade.
Fisioterapia
Fisioterapeutas podem auxiliar na recuperação da paralisia facial e tratamento do desequilíbrio e perda de força que eventualmente venham a ocorrer.
Observação ativa
Significa reavaliar periodicamente os sintomas clínicos, em especial a função auditiva e a visão (risco de catarata) e o tamanho dos schwannomas. Esta parece ser a melhor conduta para a maioria das pessoas com NF2-SWN. A observação ativa pode variar de alguns anos a poucos meses sem qualquer tratamento cirúrgico ou quimioterápico.
Var abaixo o fluxograma da observação ativa e outras condutas, considerando a perda auditiva e o tamanho do schwannoma. Adaptamos o resumo das recomendações do Consenso de 2012 e de 2020 na Figura 3 abaixo.
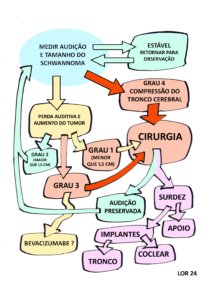
Figura 3 – Fluxograma da observação ativa e outras condutas para os schwannomas vestibulares nas pessoas com NF2-SWN. Adaptado dos Consensos de 2012 e 2020. Observação: Nos manuais de cirurgia recomenda-se cirurgia em um dos tumores bilaterais no Grau 1 com a intenção de preservar a audição, mas não conseguimos dados mostrando este desfecho favorável.
Ressecção cirúrgica
A cirurgia (ressecção parcial ou total) de um schwannoma deve ser sugerida à pessoa com NF2-SWN e sua família quando:
- O tumor ameaçar a vida (Grau 4 da classificação de Roos).
- Houver piora clínica (audição e outros sintomas) desde a última consulta.
- Seja mais provável a preservação da audição do que sua perda (ver fluxograma acima).
- Tumores menores do que 1,5 cm são candidatos à primeira cirurgia.
- A pessoa esteja consciente de que o risco pós-operatório de paralisia facial é de cerca de 30%.
- A pessoa esteja consciente de que o risco de zumbido pós-operatório é de cerca de 40%.
- A monitorização contínua do nervo facial seja garantida durante a cirurgia ([1]).
- A equipe cirúrgica tenha experiência com schwannomas vestibulares em pessoas com NF2-SWN.
Radio cirurgia e radioterapia
São ambos tratamentos contraindicados nas pessoas com NF2-SWN por risco de transformação maligna. No entanto, esta técnica tem sido indicada nas pessoas com schwannomas esporádicos (sem NF2-SWN) menores do que 3 cm. As condições e habilitação da equipe cirúrgica podem entrar no cálculo dos riscos e benefícios.
Medicamentos
Até o presente momento, apenas o medicamento bevacizumabe tem se mostrado útil em casos selecionados de schwannomas vestibulares em pessoas com NF2-SWN (ver aqui comentário detalhado sobre este medicamento).
Aparelhos auditivos
Comuns
Os aparelhos auditivos comuns (usados nas pessoas idosas, por exemplo), que apenas aumentam a intensidade do estímulo sonoro em determinadas frequências, não funcionam adequadamente nas pessoas com NF2-SWN.
Implantes
Quando os tumores ou o resultado das cirurgias resultaram em surdez completa, aparelhos auditivos especiais podem ser implantados na cóclea (se houver nervo coclear funcional) ou no tronco cerebral. Estas possibilidades podem ser compreendidas e discutidas com especialistas a partir desta revisão científica de 2016: “Restauração da audição em pacientes com NF2” (ver aqui artigo completo em inglês).
Dúvidas e sugestões
Estas informações podem ser melhoradas com sua ajuda.
Envie-nos suas dúvidas, sugestões e críticas para rodrigues.loc@gmail.com
Assinam este texto:
Profa. Dra. Juliana Ferreira de Souza
Professora do Departamento de Pediatria da Faculdade de Medicina da Universidade Federal de Minas Gerais – Coordenadora do Centro de Referência em Neurofibromatoses do Hospital das Clínicas da UFMG
Dra. Luíza Cançado Guerra D’Assumpção
Neurocirurgiã com Mestrado e Doutoranda em Neurocirurgia na Universidade Federal de Minas Gerais
Prof. Dr. Luiz Oswaldo Carneiro Rodrigues
Professor Aposentado da UFMG e Diretor Administrativo da Associação Mineira de Apoio aos Portadores de Neurofibromatose (AMANF)
Belo Horizonte, janeiro de 2024
Agradecemos as leituras e as ótimas sugestões de: Ramon Cosenza, Thalma de Oliveira Rodrigues, Fabíola A.A. Rocha Bastos, Érica Onofre e Aline da Silva Freitas.
[1] Importante ressaltar que a abordagem cirúrgica deve acontecer com monitoramento tanto nos schwannomas nos pares cranianos que possam ser afetados pela cirurgia quanto na coluna, permitindo à equipe decidir a melhor abordagem, se deve remover todo o tumor ou manter parte dele para preservar os nervos ao redor.