
Somos médicas e médicos no CRNF que acompanham centenas de famílias com Neurofibromatose do Tipo 1 (NF1), uma doença que pode causar neurofibromas. Esses tumores são geralmente benignos, mas podem trazer dor, deformidades e outras complicações. Por isso, estamos sempre em busca de tratamentos que possam controlar ou reduzir esses tumores.
Além disso, em nossa equipe no CRNF, somos 3 médicos e uma médica que têm filhas ou filhos com NF1, portanto, foi com grande interesse que lemos o estudo recente de Chen e colaboradores, publicado na revista científica The Lancet [ver aqui link para o artigo completo em inglês), que avaliou o uso do selumetinibe em adultos com neurofibromas sintomáticos que não podem ser tratados com cirurgia.
Hoje, a cirurgia é o tratamento padrão para alguns casos, mas em tumores muito grandes, espalhados ou em locais difíceis, a cirurgia não é viável. Nesses casos, não há muitos tratamentos disponíveis, o que torna esse tipo de pesquisa especialmente importante.
Uma nova possibilidade de tratamento
O selumetinibe pertence a uma classe de medicamentos chamados inibidores de MEK, que foi a primeira a mostrar alguma redução no tamanho dos neurofibromas. Esse remédio não é uma cura, e tem limitações quanto à eficácia e aos efeitos colaterais, mas representa um avanço. Até agora, ele tinha sido estudado apenas em crianças [ver aqui e aqui], e este estudo traz informações importantes sobre seu uso em adultos.
Resultados principais do estudo
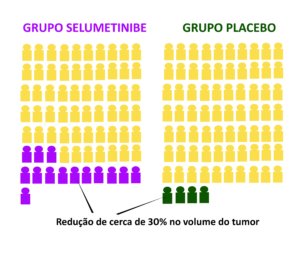
O estudo foi feito em vários centros e envolveu 145 adultos com NF1 e ao menos um neurofibroma plexiforme sintomático e inoperável. Os resultados mostraram que, após 16 ciclos de tratamento, 20% dos pacientes que tomaram selumetinibe tiveram uma redução de pelo menos 34% no volume do tumor. No grupo que tomou placebo (um remédio sem efeito), apenas 5% tiveram alguma redução.
O selumetinibe não foi superior ao placebo na redução da dor, porque houve uma pequena melhora na dor em quem usou o selumetinibe, mas esta redução da dor foi semelhante em quem usou o placebo.
No entanto, o selumetinibe não melhorou a qualidade de vida dos pacientes em comparação com o placebo.
Quais tipos de neurofibromas responderam melhor?
Existem diferentes tipos de neurofibromas: cutâneos (na pele), nodulares e difusos (ver aqui mais detalhes). Esses dois últimos podem formar tumores mais complexos chamados plexiformes, e às vezes afetam todos os nervos que saem da coluna (forma espinhal da NF1).
O estudo não informou se algum desses subtipos respondeu melhor ao tratamento com selumetinibe. Essa informação seria muito útil para orientar melhor os médicos e pacientes sobre quais pessoas têm maior chance de responder melhor ao medicamento.
Por que não foi feito um estudo com troca de grupos?
É uma pena que o estudo não tenha usado um tipo de pesquisa em que TODOS os participantes trocam de tratamento após um tempo (por exemplo, quem começou com placebo passa para o selumetinibe, e vice-versa), o que é comum em estudos com medicamentos de uso crônico.
Só houve uma troca dos pacientes que estavam usando placebo e passaram a usar o selumetinibe.
A falta deste cruzamento limita a utilidade deste estudo da Lancet pois poderíamos ficar sabendo o que acontece quando o tratamento é interrompido: o tumor volta a crescer? O efeito se mantém?
Tolerância ao tratamento em adultos com NF1 de longa duração
Os efeitos colaterais mais comuns do selumetinibe foram problemas de pele, intestinais e no fígado. Cerca de 15% dos pacientes pararam o tratamento por causa desses efeitos. Além disso, o remédio exige monitoramento constante, como exames cardíacos, o que é um desafio, especialmente porque estamos lidando com uma doença crônica e em princípio benigna.
Como os tumores da NF1 geralmente não são malignos, e o remédio só reduz parcialmente o tamanho, é importante pensar com cuidado sobre os riscos e benefícios no longo prazo. Os pacientes devem participar ativamente dessa decisão, e o tratamento deve ser feito com acompanhamento cuidadoso, como ocorre em centros especializados, como o Centro de Referência em Neurofibromatose da UFMG [ver aqui].
Todas as pessoas que precisarem podem ter acesso ao medicamento?
Também lamentamos que o estudo não tenha avaliado se o tratamento é viável do ponto de vista econômico nos adultos. Já sabemos que, para crianças, alguns estudos sugerem que o remédio só seria custo-efetivo se tivesse o preço bastante reduzido ou se o tratamento fosse curto [referências NICE e CADTH]. Mas essa análise ainda falta para os adultos.
Conclusão
O estudo de Chen et al. representa um avanço importante na pesquisa sobre tratamentos para NF1 em adultos. O selumetinibe pode ajudar uma parte dos pacientes com tumores inoperáveis, mas os benefícios ainda são limitados. Há necessidade de mais estudos para:
- Identificar melhor quem tem mais chance de se beneficiar do remédio;
- Entender quais tipos de tumores respondem melhor (nodulares ou difusos);
- Testar combinações de medicamentos diferentes;
- Avaliar os efeitos no longo prazo – o que acontece quando o medicamento é suspenso?
- E verificar se o tratamento é viável do ponto de vista econômico para que todas as pessoas que necessitem dele tenham acesso.
Continuaremos acompanhando os avanços da ciência e informando nossas famílias com transparência e responsabilidade.
Assinam este post a Diretoria da AMANF e a equipe médica do CRNF HC UFMG


SIM, somos a favor da incorporação do selumetinibe pelo SUS, mas com algumas condições, que são importantes para garantir o acesso e a segurança, para quem precisa do remédio, e a sustentabilidade do sistema de saúde:
- Redução do preço em cerca de 90% do preço já listado na CMED (como o Canadá e o Reino Unido conseguiram – ver aqui o link) para que todas as pessoas com NF1 com indicação para seu uso tenham acesso ao medicamento.
- Seja prescrito dentro da bula, ou seja, somente para neurofibromas plexiformes sintomáticos e inoperáveis em crianças de 2 a 18 anos, conforme os pacientes incluídos no estudo original (ver aqui);
- Seja prescrito por profissionais com experiência em neurofibromatoses, sem conflitos de interesse;
- Que seja seguido um protocolo de cuidados que apoie a decisão compartilhada para a decisão do uso do medicamento, que informe claramente aos pacientes os potenciais benefícios e riscos do tratamento (ver aqui os cuidados página da AMANF);
- Que todos os pacientes sejam informados claramente de que ainda não se sabe o que acontece se tivermos que suspender o selumetinibe, inclusive de que há relatos de casos de crescimento acelerado dos tumores com a interrupção do medicamento e isso é um risco a ser considerado (ver aqui relato de caso com o efeito rebote);
- Somente continuar o uso depois de 18 meses de tratamento se houver evidências clínicas objetivas de que o paciente está respondendo ao medicamento, incluindo avaliação objetiva de redução da dor, da melhora da capacidade motora e/ou da redução do tamanho do tumor.
Assinam esta opinião a equipe de médicas e médicos do CRNF e toda a Diretoria da AMANF

Convidei a Dra. Pollyanna Barros Batista para comentar um dos temas livres do Congresso de Neurofibromatose em Washington, que se encerrou na semana passada. Dra. Pollyanna participa do CRNF há mais de uma década, contribuindo com diversos conhecimentos originais sobre os problemas fonoaudiológicos na NF1 e nas Schwannomatoses.
Dr. Lor
Pollyanna Barros Batista
Fonoaudióloga
Doutora e Mestre em Ciências Aplicadas à Saúde do Adulto
Especialista em Linguagem pelo Conselho Federal de Fonoaudiologia
Idealizadora da Plataforma Hey Ear
Durante a NF Conference 2025, um dos pôsteres que mais chamou minha atenção foi o trabalho da Dra. Anna Wild e sua equipe da Universidade de Manchester, intitulado “Examining the Effect of Non-Invasive Brain Stimulation on Working Memory in NF1”. Trata-se de uma investigação ousada, que busca compreender como dois tipos de estimulação cerebral – a transcranial direct current stimulation (tDCS) e a transcranial alternating current stimulation (tACS) – podem influenciar a memória de trabalho em adolescentes com Neurofibromatose tipo 1 (NF1).
A escolha do foco na memória de trabalho não é trivial. Sabemos que muitas pessoas com NF1 enfrentam dificuldades cognitivas importantes, sobretudo em funções executivas como atenção, memória e controle inibitório. Esses desafios impactam diretamente a aprendizagem, o comportamento e a qualidade de vida, especialmente na infância e adolescência.
O estudo avaliou 30 adolescentes entre 11 e 17 anos com NF1, submetendo-os a sessões de tDCS, tACS ou estimulação simulada (sham), enquanto realizavam tarefas cognitivas em ambiente de ressonância magnética. A pesquisa é inovadora por utilizar espectroscopia funcional (fMRS) para medir em tempo real o equilíbrio entre os sistemas excitatório (glutamato) e inibitório (GABA) no cérebro – um tema central na compreensão das alterações neurológicas da NF1. No poster, como pontos negativos, não foram citados o tempo que cada participante foi submetido ao estímulo, a intensidade da corrente elétrica quando utilizaram o tDCS e as áreas em que foram posicionados os eletrodos.
Os resultados preliminares, com metade da amostra analisada (n=15), indicam que a tACS foi mais eficaz do que a tDCS em melhorar o desempenho em uma tarefa de memória de trabalho (2-back), embora nenhum dos métodos tenha promovido ganhos sustentados após o término de 3 encontros com a estimulação.
Outro achado relevante foi o aumento da atividade glutamatérgica durante as sessões de estimulação, sugerindo que ambas as técnicas têm potencial de modular a atividade cerebral. Contudo, os autores não encontraram uma relação direta entre esses níveis bioquímicos e as mudanças no desempenho das tarefas cognitivas – o que nos mostra que os mecanismos por trás desses efeitos ainda são complexos e precisam ser melhor compreendidos.
Como fonoaudióloga atuante na área de linguagem e neurodesenvolvimento, vejo esse estudo como um passo importante no caminho da personalização das terapias para pessoas com NF1. A estimulação cerebral não-invasiva ainda é uma tecnologia em fase experimental nesse público, mas carrega consigo a promessa de ser incorporada, futuramente, a programas terapêuticos mais eficazes e direcionados.
É claro que são necessários ensaios clínicos maiores e mais robustos, como os próprios autores reconhecem. Mas o fato de estarmos investigando essas possibilidades com rigor científico já é motivo de otimismo. Estudos como esse alimentam a esperança de que, no futuro, possamos oferecer abordagens terapêuticas mais precisas, que respeitem as particularidades neurobiológicas de quem vive com NF1.