

No quarto dia da Conferência do CTF em Washington (Estados Unidos), a Dra. Juliana Souza participou de painéis e palestras e nos traz os comentários dos participantes do evento sobre a sessão de posters onde ela apresentou outro trabalho científico desenvolvido em nosso CRNF (ver aqui nos anais do Congresso os resumos em inglês).
A seguir o resumo do nosso trabalho apresentado (em português).
Título
A terapia com inibidores de MEK pode ser descontinuada com segurança em um neurofibroma plexiforme responsivo? Um relato de caso
Souza JF (M.D., Ph.D.); Rezende NA (M.D., Ph.D.), Cota BCL (M.D., Ph.D.), Rodrigues LO (M.D., M.Sci.), Rodrigues LOC (M.D., Ph.D.). Centro de Referência Ambulatorial de Neurofibromatose (CRNF), Hospital das Clínicas (HC), Universidade Federal de Minas Gerais (UFMG), Brasil, rodrigues.loc@gmail.com
Introdução
O uso de inibidores de MEK (MEKi) tem sido recomendado para o tratamento de neurofibromas plexiformes sintomáticos inoperáveis (ISPN) em indivíduos com tipo de neurofibromatose (NF1) (1,2). Seguindo nosso protocolo de atendimento clínico para terapia MEKi no tratamento de ISPN (3), iniciamos o tratamento em um paciente do sexo masculino de 9 anos (DBF) com um extenso neurofibroma nodular em formato plexiforme afetando a pelve e o membro inferior direito (Figura 1A), acompanhado de dor neuropática intratável sem resposta à terapia anterior, incluindo imatinibe. O PET-CT pré-tratamento não mostrou transformação maligna. O acesso ao selumetinibe foi concedido por ordem judicial, e a terapia oral começou a ser de 25 mg/m² , duas vezes ao dia em ciclos de 28 dias a partir de dezembro de 2023.
Objetivo deste resumo
Explorar considerações clínicas sobre a descontinuação da terapia com selumetinibe, com foco no momento mais adequado, critérios e estratégias para interromper o tratamento com segurança sem causar crescimento tumoral ou recorrência da dor neuropática, seja devido à preferência do paciente ou limitações financeiras (4).
Métodos
Os exames de ressonância magnética foram realizados a cada 6 meses, pois a análise volumétrica de ressonância magnética não estava disponível em nossa instituição. Os níveis de dor foram avaliados usando uma escala de dor padronizada e as medições da circunferência da coxa foram feitas antes e durante o tratamento (dezembro de 2023 a abril de 2025) (Ver gráfico abaixo).
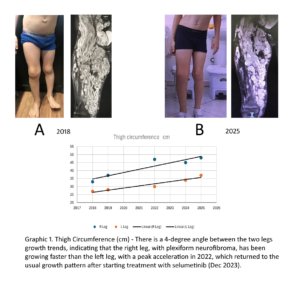
Figura 1 – Fotos dos membros inferiores de uma criança com NF1 acompanhada no CRNF por 8 anos, com ressonância magnética mostrando a extensão do neurofibroma nodular (do tipo plexiforme) e a evolução do perímetro das coxas ao longo dos anos, com e sem selumetinibe.
Resultados
Após 3 meses de tratamento, DBF relatou melhora significativa no controle da dor, uso de analgésicos não opioides de forma intermitente, e melhora na qualidade de vida, incluindo o retorno às atividades físicas na escola. Além disso, a família do paciente relatou uma percepção subjetiva de crescimento lento do ISPN, embora a ressonância magnética não tenha mostrado uma redução no tamanho do tumor (Figura 1B). O Gráfico 1 ilustra as mudanças na circunferência da coxa de 2018 a 2025. Em abril de 2025, as avaliações clínicas indicaram melhora sustentada da dor, qualidade de vida e diminuição da taxa de crescimento do tumor (Figura 1B), com eventos adversos mínimos relatados.
Conclusão
A avaliação clínica mais recente levou a uma avaliação entre a família do paciente e a equipe médica sobre os próximos passos do tratamento. Dado que o medicamento é aprovado pela FDA (e pela ANVISA no Brasil) sem protocolo prescrito para descontinuação quando as respostas ao tratamento são favoráveis, a questão de quando e como interromper a terapia permanece obscura.
Acreditamos que é hora de a comunidade NF se envolver em uma discussão completa sobre os resultados da interrupção da terapia MEKi para neurofibromas plexiformes. Esse diálogo é particularmente importante para apoiar melhor os indivíduos que podem não ser capazes de tolerar os efeitos colaterais, não podem pagar pelo tratamento ou preferem interromper a medicação.
Palavras-chave: neurofibromatose, neurofibromas plexiformes, inibidores de MEK, selumetinibe, cuidados clínicos Conflito de interesse: nenhum. Financiamento da AMANF – Associação Mineira de Apoio aos Portadores de Neurofibromatose
Referências
- Selumetinib in children with inoperable plexiform neurofibromas. Gross AM, Wolters PL, Dombi E, et al. N Engl J Med. 2020;382:1430–1442.
- Selumetinib in children with neurofibromatosis type 1 and asymptomatic inoperable plexiform neurofibroma at risk for developing tumor-related morbidity. Gross AM, Glassberg B, Wolters PL, et al. Neuro Oncol. 2022;24:1978–1988.
- Clinical Care Protocol to use selumetinib as a treatment for inoperable plexiform neurofibromas – a proposal. Rodrigues LO, Rezende NA, Cota BCL, et al. European Neurofibromatosis Meeting, 2020, Rotterdam. European Neurofibromatosis Meeting Abstracts & Presentations, 2020. https://amanf.org.br/atendimento-medico/selumetinibe/
- Challenges of selumetinib therapy for neurofibromatosis in a resource-limited setting. Khalaf T M, Alqadhi A A. 2025. Cureus 17(3): e81071.
Alguns dos comentários de participantes do evento que compareceram ao poster.
- Um médico falou sobre o que aconteceu quando ele interrompeu o uso do selumetinibe numa pessoa com glioma óptico (provavelmente uma paciente de algum estudo clínico) e houve a necessidade de trocar para a quimioterapia tradicional (no caso do glioma há esta opção, mas não nos neurofibromas): a paciente teve efeitos colaterais (dermatológicos) mais intensos após a puberdade (acne) com o selumetinibe e o glioma voltou a crescer com a interrupção da medicação.
- Outro médico, norte americano (que já trabalhou em Curitiba), conversou com colegas sobre o mesmo dilema enfrentado com um adolescente usando selumetinibe para um neurofibroma difuso mediastinal e mencionou uma proposta de redução progressiva da dose (desmame), evitando interrupção súbita. Ambos falaram de novo crescimento do tumor após a interrupção da medicação.
- A maior parte das pessoas compartilhou que este é um grande dilema que elas também têm enfrentado. Todas estão em busca de informações sobre o que acontece com a interrupção dos iMEK. Uma das médicas disse que o título do nosso pôster deveria ser “a questão de um milhão de dólares”.
- Houve relatos de pacientes, na maior parte adolescentes, que perguntam quanto tempo mais terão que usar o medicamento.
No geral, a percepção foi de que este tema é muito importante e está presente em diversos paineis, conferências e temas livres (posters) do evento.
AMANHÃ TRAREMOS A CONTINUAÇÃO DO RELATO DOS PAINEIS E CONFERÊNCIAS DA SEGUNDA FEIRA 23/06/25

No terceiro dia da Conferência do CTF em Washington (Estados Unidos), a Dra. Juliana Souza e o Dr. Bruno Cota participaram de painéis e palestras e apresentaram os dois trabalhos científicos desenvolvidos em nosso CRNF aprovados pela comissão científica (ver aqui nos anais do Congresso os resumos em inglês).
Na foto, Dra. Juliana diante do pôster do Dr. Bruno (ver abaixo o pôster mais nítido).
Apresentamos abaixo o relato da Dra. Juliana Souza, pois o Dr. Bruno Cota teve que retornar ao Brasil por problema de saúde em sua família, para quem desejamos pronta melhora.
Domingo 22/06
Minha impressão geral é de que foi um dia dedicado aos esforços atuais e às inovações que podem auxiliar no tratamento de manifestações tumorais da Neurofibromatose do tipo 1 (NF1) e Schwannomatoses (SWN), especialmente dos tumores malignos da bainha do nervo periférico (TMBNP) e schwannomas vestibulares.
- Algumas informações podem ser destacadas em estudos com modelos animais:
- As células cancerosas podem se recuperar da senescência induzida por quimioterapia e radioterapia e voltar a se dividir e crescer o tumor. A droga ABT-236 em estudo (ver resumo nos anais) pode promover a apoptose (morte da célula cancerosa).
- Foram apresentadas diferentes associações de compostos, que atuam em diferentes pontos das cascata RAS/ERK, testados in vitro e em modelos animais, com avaliação do efeito na proliferação e crescimento celular.
- A inibição da proteína do estresse térmico tipo 2 (iSHP2) no tratamento do TMBNP e a combinação do iSHP2 e CDK4 foi efetiva no modelo animal com TMBNP, parecendo conferir atividade antitumoral mais profunda e duradoura.
- A inibição da Quinase de Adesão Focal no crescimento dos schwannomas e preservação da audição está sendo estudada, assim como o Brigatinib, mencionado anteriormente.
- Inibição da via FAK tem relação com inativação dos mastócitos e pode haver sinergismo na inibição FAK e inibidores da via MEK (iMEK)
- Alguns destaques foram mencionados na prevenção do câncer foram discutidos, utilizando biomarcadores para detectar mais cedo, o que seria desejável, mas o que temos atualmente é biópsia com limitações e o PET, que é sensível mas inespecífico.Outras possibilidades apresentadas foram:
- Detectar o DNA tumoral na circulação (biópsia líquida) com múltiplas classes de potenciais marcadores.
- Menção a uma abordagem multi-ômica, ou seja, integrando a análise de DNA, RNA, proteínas e outros produtos do metabolismo celular.
- Como anda a terapia gênica na NF1?
- Pergunta-se: corrigir a expressão gênica da NF1 seria suficiente para corrigir as alterações teciduais da NF1?
- Um modelo animal está em construção, mas há limitações para a terapia gênica, como o fato de haver muitas variantes patogênicas, ser um gene grande, grande expressão na vida embrionária e outras.
- Inteligência Artificial pode ser usada na definição de biomarcadores?
- Questões sobre a habilidade de digitalizar a histopatologia;
- A possibilidade de identificar subtipos histológicos e moleculares, que podem individualizar o tratamento.
- Usou o exemplo dos gliomas de baixo grau.
- Papel do exercício físico na NF1
- Exercício voluntário atenua crescimento tumoral em modelo animal (ca de mama) – modelo animal heterozigoto
- Nf1 4,9 vezes mais chances de ca de mama
- 30% dos ca de mama têm mutação somática do gene nf1
- Atividade física de alta intensidade diminui o risco de Ca de mama em 20% ( população geral)
- No estudo apresentado por ela, animais sedentários e ativos não apresentaram alteração no peso;
- Perda do gene nf1 não modificou a composição corporal basal (há controvérsias…)
- A atividade física voluntária atrasou o surgimento dos tumores de mama e atenuaram seu crescimento
Abaixo apresentamos a versão em português do trabalho do Dr. Bruno sobre os benefícios da prática musical sobre as dificuldades cognitivas de pessoas com NF1. Amanhã apresentaremos o trabalho apresentado pela Dra. Juliana e os comentários que foram feitos sobre o pôster.
Em resumo, Dr. Bruno e sua equipe de colaboradores realizaram práticas musicais num grupo de pessoas com NF1 durante seis meses e mediram diversos indicadores cognitivos e da atividade cerebral relacionada ao processamento dos sons. Comparando os testes realizados antes e depois da prática musical, os resultados mostraram melhora significativa das funções cognitivas. Este estudo sugere que a prática musical seja útil no tratamento das pessoas com NF1.
Veja abaixo o resumo que foi apresentado em Washington (em português).

Foto do poster do Dr. Bruno exposto no CTF
Título
Efeitos da Prática Musical nas Funções Auditivas e Executivas em Indivíduos com Neurofibromatose Tipo 1
Autores: Bruno Cezar Lage Cota (PhD), Luciana Macedo Resende (PhD), Juliana Ferreira Souza (PhD), Luiz Oswaldo Carneiro Rodrigues (PhD), Julia Lemes de Medeiros, Maila Araújo Pinto, Marina Silva Corgosinho, Vitória Perlin Santiago, Jamile Noeli Andrade, Gabriela Poluceno Pereira, Thais Andreza Oliveira Barbosa, Ana Letícia Ferreira Santos de Ávila, Nilton Alves de Rezende (PhD).
Instituição: Centro de Referência em Neurofibromatose, Hospital das Clínicas, Universidade Federal de Minas Gerais, Brasil
Financiamento: AMANF
Conflitos de interesse: nenhum
Introdução
A neurofibromatose tipo 1 (NF1) é um distúrbio genético multissistêmico frequentemente associado a prejuízos nas funções executivas e no processamento auditivo, incluindo a percepção musical.
Objetivo
Examinar a relação entre déficits de função auditiva e executiva em indivíduos com NF1 e avaliar os potenciais efeitos terapêuticos da prática musical nesses domínios.
Métodos
Vinte e cinco indivíduos com NF1, com idades entre 13 e 25 anos, foram submetidos a um programa de treinamento musical de seis meses e foram avaliados pré e pós-intervenção por meio de uma bateria abrangente de testes.
O processamento auditivo foi avaliado com o teste Gaps in Noise (GIN), Pitch Pattern Sequence (PPS) e potenciais evocados auditivos de longa latência (P1-N1-P2 e Mismatch Negativity – MMN).
A percepção musical foi medida usando a Bateria de Avaliação de Amusia de Montreal (MBEA). As funções executivas foram avaliadas com o Teste de Fluência Verbal Switch (SVFT), Span de Dígitos, Tarefa de Toque de Bloco de Corsi, Teste de Cinco Dígitos (FDT), Teste de Cinco Pontos (T5P) e Teste de Rede de Atenção (ANT).
Resultados
Foram observadas correlações significativas entre as medidas musicais, auditivas e de função executiva, incluindo: MBEA e T5P (p = 0,013, r = 0,509), latência MBEA e MMN (p = 0,009, r = -0,669), GIN e Corsi (p = 0,017, r = -0,515), PPS e ANT (p = 0,006, r = -0,563) e PPS e Digit Span (p = 0,019, r = 0,506).
O treinamento musical levou a melhorias nas funções executivas, particularmente flexibilidade cognitiva (FDT: p = 0,015, η² = 0,124), troca de tarefas (FDT: p = 0,030, η² = 0,155) e fluência verbal (SVFT: p = 0,011, η² = 0,158).
O processamento auditivo também melhorou, evidenciado pelo aumento dos escores do GIN (p = 0,017, η² = 0,042) e redução da latência do MMN (p = 0,001, η² = 0,365).
Conclusão
Os achados sugerem uma forte relação entre as funções auditivas e executivas em indivíduos com NF1 e indicam que a prática musical pode ter efeitos benéficos sobre esses sistemas cognitivos, apoiando seu uso como estratégia terapêutica complementar.

Relatos da Dra. Juliana Souza e do Dr. Bruno Cota
Sábado (21/06/25)
CTF presta homenagem ao nosso amigo e pioneiro Vincent Riccardi
A homenagem póstuma ao Dr. Vincent M. Riccardi (falecido em 2024 aos 83 anos) foi um momento de grande emoção para todas as pessoas que tiveram a oportunidade de conviver com aquele que, antes de todo mundo, se envolveu profundamente com as pessoas com NF.
Riccardi esteve por duas vezes em nosso CRNF (ver aqui um desses momentos) onde deixou lições de sabedoria e espírito científico. Veja a homenagem da comunidade NF dos Estados Unidos ao Riccardi, com uma fotografia que fiz dele e de sua querida Susan no Parque Lagoa do Nado em Belo Horizonte.
Além de ser um referência internacional, estimulou nossas linhas de pesquisa e nos apoiou internacionalmente.
Vamos continuar nosso trabalho com a mesma dedicação que Riccardi nos ensinou, como uma forma de homenagear esta pessoa que tanto nos inspira.
Riccardi vive em nossos corações e mentes.
Dr. Lor
Os outros assuntos do dia foram comentados pela Dra. Juliana e Dr. Bruno foram: :
- Aula espetacular da Dra. Nancy Ratner sobre neurofibromas
- Preservação da audição nas pessoas com NF2
- Inteligência artificial – usos e limitações nas NF
- Tratamento da transformação maligna dos neurofibromas
- Como medir o volume dos neurofibromas
Confira abaixo nossos resumos
Formação dos neurofibromas, segundo a Dra. Nancy Ratner
Inicialmente, ela mostrou que a perda do segundo alelo do gene NF1 na célula de Schwann dá início aos neurofibromas (que podem ser plexiformes), mas isto não é suficiente para a transformação maligna, pois é preciso a ocorrência de outros fatores.
Em seguida, mostrou a importância dos estudos pré clínicos no avanço na utilização dos inibidos MEK (iMEK) nos estudos clínicos. Chamou a atenção para as vias SHP2 e KRAS, para as quais já há uma droga (BI-6674) em estudo pré-clínico, em preparação para publicação, assim como outros potenciais alvos.
Mostrou um diagrama com diversas drogas que tentam bloquear outras etapas da cascata e que foram capazes de encolher os neurofibromas plexiformes em modelos animais.
Relatou que a ideia atual é começar a associar estas drogas, embora ainda não haja efeito durável ou combinação que possa produzir um encolhimento maior dos neurofibromas plexiformes.
Mostrou que os neurofibromas plexiformes têm cerca de 30% de células do sistema imunitário: macrófagos, células dendríticas e células T e que tanto os iMEK quanto os iSHP2 diminuem a população de macrófagos e células dendríticas. De maneira um pouco menos dramática nos humanos do que no modelo animal, mas ainda assim importante
Destacou o fato de que as células de Schwann Nf-/Nf- expressam fatores que atraem células T e células dendríticas, enquanto os macrófagos estão dentro das células tumorais no início da sua formação e promovem seu crescimento. Drogas anti macrófagos no início da formação inibem o surgimento dos plexiformes e diminuem um pouco seu tamanho depois de formados.
Quando células B e T não estão presentes, neurofibromas plexiformes não se formam e os macrófagos estão diminuídos. Se você oferece anticorpos anti células T (ver poster de Jay Pandavela nos anais do evento), ou seja, o mecanismo seria assim: NF1(-/-) → macrófagos → citocinas → CD8 células T → neurofibromas.
Lembrou que a mesma variante patogênica tem fenótipos diferentes por causa de genes diferentes e modificadores genéticos diferentes (epigenética). Sugeriu que esta informação poderá ser útil no tratamento dos plexiformes e que já há variantes germinativas ATM germline em estudo.
Finalmente, disse que um microambiente heterozigoto não é necessário para a formação de um neurofibroma plexiforme, ao contrário do que tem sido pensado nos últimos anos.
Preservação da audição nas pessoas com Schwannomatose relacionada ao gene NF2 (NF2-SWN)
De início, o palestrante Bradley Welling apresentou uma opinião controversa, a sua preferência em operar os schwannomas vestibulares o mais precocemente possível (quando ainda tem apenas milímetros de tamanho), pois a evolução a médio e longo prazo dos implantes cocleares e de tronco cerebral não é um resultado tão satisfatório.
(Comentário nosso: ver aqui nossa sugestão de tratamento para schwannomas vestibulares nas pessoas com NF2-SWN, na qual evitamos a abordagem cirúrgica o máximo possível)
Foi ressaltada a importância de se realizar a ressecção dos schwannomas com monitorização direta do nervo coclear para preservação da audição, mas não foram apresentados dados comparativos com esse desfecho entre procedimentos com ou sem monitoração.
Sobre os mecanismos para perda auditiva na swn-nf2, foram citados o comprometimento da vasculatura, a compressão direta da cóclea pelo tumor e a secreção de proteínas citotóxicas pelo tumor.
Relatou que 75% dos pacientes com NF2-SWN não são candidatos à cirurgia com preservação auditiva no momento da cirurgia. Dos 25% que se submetem à cirurgia, 70% têm tumores menores que 1cm, e metade deles evolui com preservação da audição no pós-operatório. Contudo, pela própria evolução da doença, a taxa de preservação da audição a longo prazo não tem melhora significativa.
A discrepância entre tamanho do schwannoma e perda auditiva foi novamente destacada, estando de acordo com o protocolo proposto pelo nosso CRNF (ver acima).
Alguns fatores intra-operatórios que determinam o resultado da cirurgia para a preservação da função auditiva:
- nervo de origem anatômica do tumor (ramo coclear, nervo vestibular superior ou inferior);
- suprimento vascular da cóclea;
- expertise do cirurgião;
- extensão do tumor;
- monitorização intra-operatória da função coclear (novamente sem dados apresentados);
Como tratamento paliativo para perda da audição na NF2-SWN, apresentaram os óculos de captura de fala em tempo real, nos quais a pessoa pode ler na lente as palavras ditas no ambiente em tempo real.
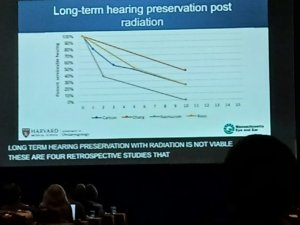
Concluiu com a apresentação de um gráfico (abaixo) sobre a preservação da função auditiva a longo prazo em diferentes trabalhos que avaliaram o efeito do tratamento dos schwannomas com radioterapia, todos de retrospectivos, mostrando que em todos os casos a audição teve piora progressiva, independente do efeito da radioterapia no tamanho do tumor.
Inteligência Artificial nas NF
Os três palestrantes do painel falaram sobre a IA e diagnósticos por imagem de tumores em oncologia, comentando as limitações para validação das ferramentos de IA, por parte dos pacientes e médicos, quanto ao seu uso na prática médica.
Existe uma IA, já disponível, que faz uma medida volumétrica em 3D dos schwannomas vestibulares na NF2-SWN. Foi questionada a capacidade de identificação pela IA de artefatos e para indicação do implante coclear.
Nos neurofibromas cutâneos ficou a impressão de que a IA teria utilidade, essencialmente, para medir a eficácia de intervenções terapêuticas.
Tratamento da transformação maligna dos neurofibromas
A Dra, Blakeley apresentou um estudo recém iniciado para tumor maligno da bainha do nervo periférico associando imunoterapia com quimioterapia com cirurgia e com radioterapia se cirurgia deixar margem com tumor.
As drogas em questão seriam:
- Nivolumabe + Ipilimumabe: imunoterapia para estudo histológico com resultado de tumor atípico ou TMBNP, cujo foco seriam PD-L1 e células T CD8 (CTLA4 e PD1);
- TMBNP recentemente diagnosticado por biópsia: 2 doses dos medicamentos nas 8 semanas entre a biópsia e o tratamento tradicional;
- Quimioterapia citotóxica pré cirurgia para melhorar as margens
- Cirurgia em busca de margem negativa.
- Se não conseguir margem negativa, radioterapia após a cirurgia.
Aguardaremos os resultados.
Análise do volume dos neurofibromas plexiformes
Este painel foi moderado por Andrea M Gross e Brigitte Widmann, ambas relacionadas aos estudos com Selumetinibe sob o patrocínio da Astrazeneca.
Foram lembradas as diversas características dos chamados neurofibromas plexiformes (de maneira generalizada), mas sem a proposta de separá-los para avaliação em nodulares, difusos e na forma espinhal.
Mostraram a diferença da medida em 1D, 2D e 3D, sendo a 3D mais precisa e, portanto, a indicada de acordo com as recomendações da “REINS Consensus Recommendation” para a medida dos neurofibromas plexiformes.
No painel mostrou-se que a definição do contorno pode ser: manual, semi automática ou por meio de IA, mas há limitações na definição de qual é o contorno para a medida do volume. A variabilidade de medida é grande entre observadores (tabela apresentada pela Eva Dombi).
Os diferentes tipos de neurofibromas plexiformes (sem distinção de difusos ou nodulares) foram mostrados como exemplos que são medidos com 3D e se apresentam para os estudos clínicos.
Foram discutidas as possibilidade sobre o que acontece quando um neurofibroma plexiforme “encolhe” com o tratamento (com iMEK, por exemplo):
- Remodelamento (a forma nodular se torna quase que o “resto” de um difuso) mas sem desaparecer;
- Mudança (diminuição) no sinal em T2 (até que ponto esta alteração modifica a definição do contorno e a medida pós iMEK? (dúvida nossa);
- Mudança na densidade do tumor.
Discutiu-se também se a precisão da medida 3D refletiria a realidade, pois há certo grau de incerteza, sendo dependente do examinador e como capta o processo de mudança. Diante disso, considerando os vieses na análise volumétrica, que uma diferença de 10% no volume deve ser considerada dentro de uma possível “margem de erro”.
Também se comentou sobre variação volumétrica e seu impacto estético: será que uma redução de 20% de volume pode representar pouco em relação a uma redução linear de um tumor (~6%)?
Nessa discussão, um dos participantes, Frank Buono (que é portador de NF2-SWN), ressaltou a relevância de se considerar a melhora dos sintomas em contraposição somente à redução do volume. Esta posição se opôs aos demais participantes do painel, envolvidos nos estudos com iMEK, que defenderam que uma diminuição de 20% no volume é um bom resultado.
Finalmente, foi debatida a relação entre a superfície dos tumores e o volume dos tumores, que pode ser extrapolada para os tumores plexiformes nodulares e difusos. Dois tumores com o mesmo volume podem ter superfícies muito diferentes (maior nos difusos), com diferente repercussão pela extensão do tumor, especialmente estética. Portanto, clinicamente, o impacto de uma pequena redução de volume (por ex, 20%) em um tumor plexiforme difuso, pode ser muito menor do que em um neurofibroma nodular.

A definição do contorno dos plexiformes sofre muitos vieses nos plexiformes difusos (mistura de tumor com tecidos sadios, estruturas normais com características de imagem semelhantes ao tumor, qualidade da imagem) impactando na variabilidade inter-examinadores.
AMANHÃ ENVIAREMOS MAIS NOTÍCIAS!!!