

Dando continuidade à divulgação online dos capítulos escritos para a Edição Comemorativa dos 20 anos do CRNF, a ser lançada em novembro de 2024, apresentamos o texto esclarecedor da Dra. Cinthia Vila Nova Santana, doutora em genética , que vem colaborando com nossos conhecimentos sobre as pessoas com NF1. Muito obrigado por mais esta colaboração, que é uma necessidade dos profissionais de saúde que trabalham com doenças genéticas.
Dr. Lor
Cinthia Vila Nova Santana
Biomédica, Doutora em Genética
Escola Bahiana de Medicina e Saúde Pública
Fundação ProAR
O desenvolvimento da engenharia genética na década de 1970 marcou o início de uma era importante na medicina. Pela primeira vez, pesquisadores foram capazes de manipular moléculas de DNA através de técnicas como a clonagem e reação em cadeia da polimerase (PCR), o que tornou possível o estudo de genes de interesse medicinal e biotecnológico. Desde então, muitos avanços aprimoraram as técnicas de biologia molecular, possibilitando o estudo dos genes não apenas isolados, mas também no organismo como um todo.
O primeiro relato de terapia gênica se deu em 1990, em uma paciente com uma deficiência imunológica grave provocada por uma condição genética rara (Anderson, 1990). Essa técnica envolvia a inserção de cópias funcionais do gene responsável pela produção de uma enzima ausente ou defeituosa. Embora esse tratamento tenha mostrado resultados promissores, também revelou desafios significativos, como a possibilidade de reações adversas, eficácia limitada e, até mesmo, risco de morte.
Anos mais tarde, as pesquisadoras Jennifer Doudna e Emmanuelle Charpentier anunciaram, em 2012, uma nova técnica de edição de genomas (CRISPR) que iria revolucionar o campo da terapia gênica. Essa descoberta conferiu, em 2020, o prêmio Nobel em Química para essas duas pesquisadoras (The Nobel Foundation, 2020). A nova metodologia reacendeu a esperança de cura para doenças genéticas, como as neurofibromatoses, que são provocadas por erros em um local específico no nosso DNA. Nesse capítulo, vamos entender um pouco mais sobre terapia gênica e como o método pode influenciar no tratamento da Neurofibromatose do tipo 1 (NF1).
Tratamentos genéticos e suas possibilidades na NF1
Antes de falarmos sobre tratamentos genéticos (ou terapia gênica), vamos compreender alguns conceitos. O nosso DNA contém genes onde estão armazenadas as instruções para a fabricação de proteínas. É como se os genes fossem o manual de instruções para a montagem de peças ─ as proteínas ─ e essas peças fazem o trabalho necessário para garantir o bom funcionamento do nosso corpo. Quando alguma instrução está defeituosa, temos proteínas que não funcionam corretamente, ou até mesmo que não são nem produzidas, e isso pode acarretar doenças genéticas como, por exemplo, a fibrose cística, anemia falciforme, beta talassemia, e as neurofibromatoses. Os tratamentos genéticos, por sua vez, têm como objetivo corrigir esses erros no DNA (também chamados de mutações), a fim de tratar uma doença (Anguela & High, 2019).
A NF1 é uma doença genética, resultante da mutação no gene que produz a proteína neurofibromina, presente em vários tipos celulares (especialmente em neurônios e nervos periféricos) e tem como função controlar o crescimento das células. Nas pessoas com NF1, essa proteína está ausente ou defeituosa, o que gera uma proliferação e um crescimento celular descontrolado, característico dos tumores (neurofibromas) observados na doença (Brems et al., 2009; Gutmann et al., 2017). Até o momento, não existe cura para a NF1, apenas tratamentos das condições específicas que surgem em cada paciente.
Mas, então, você poderia se perguntar: se o problema está na ausência da neurofibromina, por que não tomamos a neurofibromina artificial para correção da NF1? Existem alguns fatores a se considerar.
Primeiramente, esta é uma proteína grande (com cerca de 327 kDa e 2818 aminoácidos) e com uma conformação complexa (Nordlund et al., 1993; Upadhyaya et al., 2008), o que dificulta a sua síntese em laboratório.
Além disso, essa proteína é importante desde a vida intrauterina para o desenvolvimento do bebê, por isso muitas manifestações clínicas da doença, como as manchas café com leite, por exemplo, já estão presentes ao nascimento. Assim, a administração da neurofibromina após o nascimento não seria capaz de reverter as consequências da ausência dessa proteína em estágios iniciais do desenvolvimento.
Somando-se a isso, cerca de metade dos casos de NF1 são resultantes de mutações novas, quando não há histórico familiar (Mckeever et al., 2008), e, portanto, o diagnóstico da doença não é conhecido durante a gestação, o que inviabilizaria um possível tratamento.
Nesse contexto, terapias genéticas surgem como uma esperança de tratamento, e quem sabe até mesmo cura, para a NF1. Vejamos abaixo alguns dos avanços e pesquisas que estão sendo realizadas nessa área.
Terapia gênica direta – Substituição do gene defeituoso
A terapia gênica direta para NF1 busca corrigir a mutação ou substituir o gene NF1 defeituoso por uma cópia funcional (Leier et al., 2020), visando restaurar a função da neurofibromina e reduzir o crescimento dos tumores e outras manifestações associadas à doença. Essa parece ser uma excelente alternativa de tratamento, no entanto, alguns desafios ainda precisam ser superados para que a substituição do gene seja uma realidade.
Como já vimos, o NF1 é um gene grande, com cerca de 350 kb, e complexo (Pasmant et al., 2015), o que dificulta consideravelmente sua manipulação em laboratório. Pesquisadores conseguiram menos de 10% de sucesso nas taxas de transfecção, isto é, incorporação do gene funcional dentro das células (Bai et al., 2019). Além disso, o sistema de entrega do gene funcional ainda não está bem estabelecido para a NF1. Opções de entrega como a utilização de vetores virais, nanopartículas, exosomos e polímeros ainda não conseguem acomodar o tamanho do gene da NF1 em seu sistema. Imagine tentar transportar um bolo de casamento de 7 andares em uma bicicleta, é muito difícil conseguir entregar o bolo íntegro no local da festa.
CRISPR – Edição do genoma
Alternativamente à substituição do gene, a edição de parte do genoma se torna uma opção mais plausível no caso da NF1. Existem três abordagens principais para edição do DNA:
- utilização de nucleases dedo de zinco (ZFN, do inglês zinc finger nucleases);
- uso de nucleases efetoras semelhantes a ativadores de transcrição (TALENs, do inglês transcription activator-like effector nucleases);
- sistema CRISPR-Cas (do inglês Clustered Regularly Interspaced Short Palindromic Repeats).
As duas primeiras utilizam como domínio de reconhecimento do DNA estruturas proteicas (dedos de zinco e proteínas TAL – transcription activator-like, respectivamente), ao passo que o sistema CRISPR-Cas utiliza o pareamento das bases nitrogenadas (adenina com timina e citosina com guanina), o que confere maior especificidade a essa metodologia.
Desde que foi descrito como ferramenta de edição de genoma em 2012 (Jinek et al., 2012), o sistema CRISPR-Cas vem sendo amplamente utilizado devido à sua simplicidade, eficácia e versatilidade (Wang & Doudna, 2023). Vamos entender um pouco mais sobre ele.
CRISPR acontece naturalmente em bactérias, como um mecanismo de defesa contra infecções por vírus (Figura 1) (Mojica et al., 2005). De maneira geral, quando um vírus infecta uma bactéria, ele injeta seu material genético dentro dela. Parte desse material é então incorporado no DNA da bactéria como um novo espaçador do sistema CRISPR. Vamos imaginar que a bactéria é uma casa que foi invadida pelo vírus. As câmeras de segurança conseguem identificar esse vírus e guardar a sua imagem nos arquivos (espaçadores do sistema CRISPR). Essa imagem é impressa e entregue aos seguranças para que fiquem alertas em caso de nova tentativa de invasão pelo vírus. No caso da bactéria, a sequência do vírus é, então, transcrita (“impressa”, isto é, a informação é passada de DNA para RNA) e processada para gerar os RNAs de CRISPR (crRNAs), como observado na Figura 1. Estes, por sua vez, associam-se a uma proteína Cas, que irá cortar o material genético do vírus invasor utilizando o crRNA como molde para esse silenciamento em infecções futuras (Wang & Doudna, 2023).
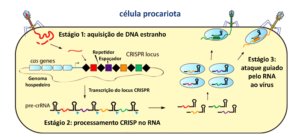
Figura 1: Representação esquemática do sistema CRISPR numa célula procariota. Com uma infecção por vírus, a bactéria incorpora parte desse material genético em seu próprio genoma, como um espaçador do sistema CRISPR (estágio 1). Essa informação será transformada (transcrita) em um pré-crRNA (estágio 2) e então processada para que, em caso de uma nova infecção viral, o crRNA maduro possa clivar (destruir) o material genético do vírus (estágio 3), impedindo o sucesso da infecção.
Inspirados nesse sistema, os pesquisadores começaram a trabalhar na possibilidade de utilizar CRISPR como ferramenta para edição de genomas. Após anos de estudo, o grupo liderado por Jennifer Doudna e Emmanuelle Charpentier descobriu como realizar esse feito que iria revolucionar o campo das ciências biomédicas. Eles identificaram que para editar o genoma com CRISPR eram necessários basicamente três componentes (Figura 2) (Jinek et al., 2012):
- RNA guia: é a combinação do crRNA (onde está a sequência complementar ao alvo) com o tracrRNA (contribui para a estabilidade da estrutura e seu reconhecimento pela enzima Cas);
- Enzima Cas (do inglês CRISPR associated): essa enzima possui a capacidade de cortar o DNA na região específica identificada pelo RNA guia. Existem várias enzimas descritas hoje em dia, mas a primeira delas foi a Cas9;
- DNA alvo com a sequência PAM: o DNA alvo é a sequência de interesse que será editada. Próximo a ela, está o motivo adjacente ao protoespaçador ou PAM (do inglês, Protospacer Adjacent Motif), que é uma sequência curta (cerca de 2 a 5 nucleotídeos) essencial para a ancoragem do complexo Cas/RNA guia. A sequência PAM funciona como uma bandeira para confirmação que aquela região do DNA alvo é realmente onde deve acontecer a edição.
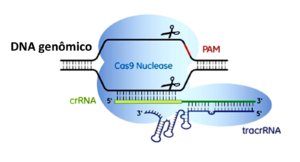
Figura 2: Representação esquemática do sistema CRISPR como ferramenta de edição do genoma. Basicamente são necessários apenas três componentes, o RNA guia formado pela combinação do crRNA e o tracrRNA, a enzima Cas9 e o DNA alvo juntamente com a sequência PAM.
De maneira geral, o que acontece na edição do genoma é a entrega do complexo contendo o RNA guia acoplado à enzima Cas, para o tecido que necessita da edição. Um dos métodos mais comuns de entrega é através de vetores virais, que consistem em vírus modificados para não causarem doença, assim como por nanopartículas, exosomos, eletroporação, entre outros métodos de transporte.
Diversas adaptações foram realizadas nesse sistema para permitir uma atuação mais ampla na edição do genoma. Por exemplo, é possível modificar uma base nitrogenada sem necessariamente clivar o DNA utilizando proteínas acopladas à Cas, ou ativar e desativar genes, regular a expressão de genes através de modulação epigenética (afrouxando ou condensando o DNA para deixá-lo mais ou menos disponível para a maquinaria de transcrição da célula), entre outras aplicações [para maiores detalhes, ver revisão de (Chavez et al., 2023)].
Inúmeros estudos utilizam o sistema CRISPR como ferramenta de edição genética na busca pelo tratamento de doenças, mas, até o momento, apenas uma terapia gênica com essa ferramenta foi aprovada para uso em pacientes. No final de 2023, o Reino Unido, seguido dos EUA, aprovaram a terapia com CRISPR para tratamento da anemia falciforme e beta talassemia (Food and Drug Administration (FDA), 2023), após anos de pesquisas e acompanhamento de pacientes. Os resultados são bastante promissores e nos dão esperança para que, em breve, essa metodologia possa ser empregada no tratamento de outras doenças.
No que se refere à NF1, ainda existem poucos estudos com essa metodologia. De maneira geral, a literatura mostra a utilização da ferramenta CRISPR para criar organismos modelos experimentais da doença, a fim de que pesquisas mais aprofundadas possam ser realizadas para melhor entendimento da doença e busca de alvos terapêuticos. Contudo, até então, não existem publicações com o uso de CRISPR no tratamento da NF1.
Afinal, existe cura da NF1 com terapia gênica?
Leier e colaboradores e Staedtke e colaboradores resumem, em seus artigos de revisão, possibilidades de terapias genéticas para a NF1 (Leier et al., 2020; Staedtke et al., 2024). A terapia gênica é sim um grande avanço na medicina, no entanto, existem algumas limitações. É importante destacar que o tamanho e complexidade do gene NF1, a dificuldade com o sistema de entrega (vetores virais e não virais), o aspecto sistêmico da doença que afeta múltiplos órgãos e tecidos, a importância da neurofibromina desde a vida uterina, o diagnóstico tardio e a alta taxa de mutações novas em pessoas sem histórico familiar são alguns dos fatores que dificultam os avanços para o tratamento genético da NF1. Apesar disso, a ciência está caminhando em busca desse tratamento e espera-se que, em breve, novas possibilidades se tornem realidade.
Referências
Anderson, W. F. (1990). September 14, 1990: The Beginning. Human Gene Therapy, 1(4), 371–372. https://doi.org/10.1089/hum.1990.1.4-371
Anguela, X. M., & High, K. A. (2019). Entering the Modern Era of Gene Therapy. Annual Review of Medicine, 70(1), 273–288. https://doi.org/10.1146/annurev-med-012017-043332
Bai, R. Y., Esposito, D., Tam, A. J., McCormick, F., Riggins, G. J., Wade Clapp, D., & Staedtke, V. (2019). Feasibility of using NF1-GRD and AAV for gene replacement therapy in NF1-associated tumors. Gene Therapy, 26(6), 277–286. https://doi.org/10.1038/s41434-019-0080-9
Brems, H., Beert, E., de Ravel, T., & Legius, E. (2009). Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. The Lancet Oncology, 10(5), 508–515. https://doi.org/10.1016/S1470-2045(09)70033-6
Chavez, M., Chen, X., Finn, P. B., & Qi, L. S. (2023). Advances in CRISPR therapeutics. In Nature Reviews Nephrology (Vol. 19, Issue 1, pp. 9–22). Nature Research. https://doi.org/10.1038/s41581-022-00636-2
Food and Drug Administration (FDA). (2023, December 8). FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. Https://Www.Fda.Gov/News-Events/Press-Announcements/Fda-Approves-First-Gene-Therapies-Treat-Patients-Sickle-Cell-Disease.
Gutmann, D. H., Ferner, R. E., Listernick, R. H., Korf, B. R., Wolters, P. L., & Johnson, K. J. (2017). Neurofibromatosis type 1. Nature Reviews Disease Primers, 3, 17004. https://doi.org/10.1038/nrdp.2017.4
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., & Charpentier, E. (2012). A Programmable Dual-RNA – Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (New York, N.Y.), 337(August), 816–822. https://doi.org/10.1126/science.1225829
Leier, A., Bedwell, D. M., Chen, A. T., Dickson, G., Keeling, K. M., Kesterson, R. A., Korf, B. R., Marquez Lago, T. T., Müller, U. F., Popplewell, L., Zhou, J., & Wallis, D. (2020). Mutation-Directed Therapeutics for Neurofibromatosis Type I. In Molecular Therapy Nucleic Acids (Vol. 20, pp. 739–753). Cell Press. https://doi.org/10.1016/j.omtn.2020.04.012
Mckeever, D., Mckeever, K., Shepherd, C. W., Crawford, H., & Morrison, P. J. (2008). An epidemiological, clinical and genetic survey of Neurofibromatosis type 1 in children under sixteen years of age. In Ulster Med J (Vol. 77, Issue 3). www.ums.ac.uk
Mojica, F. J. M., Díez-Villaseñor, C., García-Martínez, J., & Soria, E. (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of Molecular Evolution, 60(2), 174–182. https://doi.org/10.1007/s00239-004-0046-3
Nordlund, M., Gu, X., Shipley, M. T., & Ratner, N. (1993). Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 13(4), 1588–1600. http://www.ncbi.nlm.nih.gov/pubmed/8463837
Pasmant, E., Parfait, B., Luscan, A., Goussard, P., Briand-Suleau, A., Laurendeau, I., Fouveaut, C., Leroy, C., Montadert, A., Wolkenstein, P., Vidaud, M., & Vidaud, D. (2015). Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations? European Journal of Human Genetics, 23(5), 596–601. https://doi.org/10.1038/ejhg.2014.145
Staedtke, V., Anstett, K., Bedwell, D., Giovannini, M., Keeling, K., Kesterson, R., Kim, Y. R., Korf, B., Leier, A., McManus, M. L., Sarnoff, H., Vitte, J., Walker, J. A., Plotkin, S. R., & Wallis, D. (2024). Gene-targeted therapy for neurofibromatosis and schwannomatosis: The path to clinical trials. Clinical Trials, 21(1), 51–66. https://doi.org/10.1177/17407745231207970
The Nobel Foundation. (2020, October 7). The Nobel Prize. Https://Www.Nobelprize.Org/Prizes/Chemistry/2020/Press-Release/.
Upadhyaya, M., Kluwe, L., Spurlock, G., Monem, B., Majounie, E., Mantripragada, K., Ruggieri, M., Chuzhanova, N., Evans, D., Ferner, R., Thomas, N., Guha, A., & Mautner, V. (2008). Germline and Somatic NF1 Gene Mutation Spectrum in NF1-Associated Malignant Peripheral Nerve Sheath Tumors (MPNSTs). Human Mutation, 29, 74–82. https://doi.org/10.1002/humu
Wang, J. Y., & Doudna, J. A. (2023). CRISPR technology: A decade of genome editing is only the beginning. Science, 379(6629). https://doi.org/10.1126/science.add8643
Outras informações sobre este tema em nosso site:
https://amanf.org.br/2016/04/crispr-nf/
https://amanf.org.br/2015/12/selecionar-bebes-sem-nf/
https://amanf.org.br/2016/02/novidade-no-dna-das-pessoas-com-nf1/
https://amanf.org.br/2016/03/pergunta-188-por-que-nao-dar-neurofibromina-para-quem-tem-nf1/
https://amanf.org.br/2016/09/reproducao-assistida/
https://amanf.org.br/2018/08/novas-mutacoes/
https://amanf.org.br/2019/04/quando-sera-cura-neurofibromatoses/





 Participaram da banca examinadora os professores (na foto, da esquerda para a direita) Maria Raquel Santos Carvalho (UFMG), Juliana Ferreira de Souza (do Centro de Referência em Neurofibromatoses e do Centro Universitário UNIBH), Marcelo Rizzatti Luizon (UFMG), Wagner Carlos Santos Magalhães (do Instituto Mário Penna) e Renan Pedra de Souza (UFMG).
Participaram da banca examinadora os professores (na foto, da esquerda para a direita) Maria Raquel Santos Carvalho (UFMG), Juliana Ferreira de Souza (do Centro de Referência em Neurofibromatoses e do Centro Universitário UNIBH), Marcelo Rizzatti Luizon (UFMG), Wagner Carlos Santos Magalhães (do Instituto Mário Penna) e Renan Pedra de Souza (UFMG). Segundo, porque vi a dedicação da Cinthia com o projeto e seu desejo de continuar estudando as neurofibromatoses no seu regresso para a Universidade Federal da Bahia, dando-me a impressão de que seu contato conosco frutificará em novas pesquisas importantes. Além disso, seus orientadores e os demais professores da banca mostraram grande interesse pelo tema, sinalizando que novos alunos de pós-graduação interessados em problemas das neurofibromatoses podem surgir no campo da genética, dando continuidade a esta linha de pesquisa.
Segundo, porque vi a dedicação da Cinthia com o projeto e seu desejo de continuar estudando as neurofibromatoses no seu regresso para a Universidade Federal da Bahia, dando-me a impressão de que seu contato conosco frutificará em novas pesquisas importantes. Além disso, seus orientadores e os demais professores da banca mostraram grande interesse pelo tema, sinalizando que novos alunos de pós-graduação interessados em problemas das neurofibromatoses podem surgir no campo da genética, dando continuidade a esta linha de pesquisa.



