Quando tratar os neurofibromas plexiformes?
Uma das perguntas que recebemos em nossos atendimentos clínicos é:
-
Quando devemos tratar um neurofibroma plexiforme?
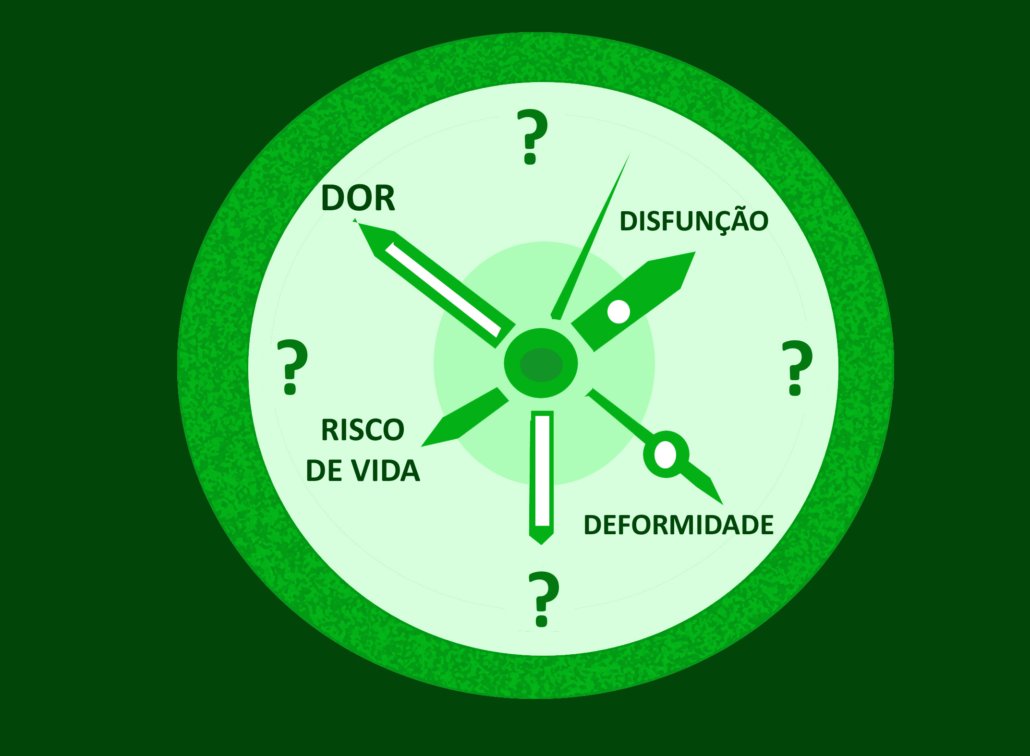
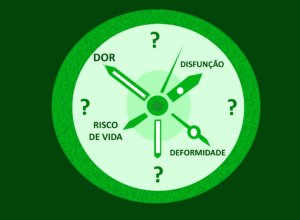
No momento, temos apenas uma regra geral: quando houver suspeita de transformação maligna (dor forte, disfunção neurológica, crescimento acelerado e endurecimento) os neurofibromas plexiformes (NP) devem ser removidos cirurgicamente (ver aqui mais informações sobre essa possibilidade).
Além dessa orientação acima, temos dificuldade para tomar a decisão de quando (e como) tratar os NP porque em todo o mundo ainda não há uma regra geral que possa ser aplicada com segurança, porque não sabemos exatamente como evoluem os NP. Quer dizer, ainda não conhecemos bem a história natural dos NP:
- quando e quanto eles vão crescer?
- quando podem dar sintomas?
- quando se transformam em tumores malignos?
Recentemente, uma equipe de cientistas da Holanda tentou responder a estas perguntas revendo os prontuários de 10 anos de acompanhamento clínico de 90 pessoas com Neurofibromatose do tipo 1 (NF1) e neurofibromas plexiformes (NP). Os resultados foram publicados na revista Cancers (ver aqui artigo completo em inglês).
Apresento abaixo o resumo adaptado do estudo (entre parênteses uso palavras menos técnicas para facilitar a leitura).
Justificativa para o estudo
Os NP são tumores benignos (no exame histológico) da bainha dos nervos periféricos associados à NF1 e muitas vezes levam à morbidade significativa (complicações que reduzem a qualidade de vida) devido ao crescimento. O tratamento inclui espera vigilante (observação atenta), cirurgia para redução parcial e, recentemente, tratamento sistêmico com inibidores de MEK (como o selumetinibe, um quimioterápico novo aprovado pela ANVISA).
No entanto, devido à escassez de estudos de história natural, a nossa compreensão da progressão natural dos NP para orientar os médicos na decisão de quem e quando intervir é insuficiente.
Este estudo teve como objetivo descrever as características dos pacientes com NF1 com NP e comparar as pessoas com alto risco de progressão do NP ou com morbidade significativa por NP (chamados de NP complexa) com as pessoas com NF1 com NPs de menor complexidade.
Métodos
Foi um estudo retrospectivo utilizando dados clínicos de registros hospitalares de pacientes com NF1 com NP atendidos no Hospital Infantil Sophia, na Holanda, entre 2012 e 2023. Foram avaliados os fenótipos clínicos (como a doença se apresenta em cada pessoa) e características dos NP que pudessem prever sua evolução, incluindo sua complexidade e o momento em que houve uma intervenção (cirúrgica ou clínica). Os resultados foram submetidos a testes estatísticos apropriados.
Resultados
Foram incluídos 90 pacientes com idade mediana na última avaliação de 15,7 anos e duração mediana de acompanhamento de 9,8 anos. Dos 90 indivíduos com NP benigna, 37 desenvolveram morbidade por NP durante o acompanhamento.
A morbidade dos NP aumentou de acordo com o passar do tempo, ou seja, quanto maior a idade da criança ou adolescente, mais morbidade foi encontrada, especialmente na cabeça e tronco.
Não houve efeito do tipo de variante genética sobre a morbidade.
Conclusão
Esse estudo piloto mostrou que o aumento da idade e a localização do NP estão associados a uma maior chance de morbidade e maior risco de intervenção. Isto pode contribuir para decisões sobre em quem e quando iniciar o tratamento em pacientes com NF1 com NP.
Meus comentários
O estudo trouxe algumas informações que precisamos analisar.
- Entre as 90 crianças e adolescentes com NP, 37 delas (41%) apresentaram alguma complicação que motivou o tratamento pela equipe médica. Ou seja, a maioria dos NP não apresentou grandes problemas durante 10 anos de acompanhamento.
- A conclusão dos autores é que quanto maior a idade da criança maior a chance de receber a indicação de tratamento. Mas isto seria mesmo o esperado, pois a NF1 é sabidamente uma doença progressiva (ver aqui). Então, à medida que o tempo passa, maior a chance de surgir alguma complicação que necessite tratamento.
- É uma pena que o estudo não tenha medido o volume dos NP por meio da ressonância magnética porque eles não tinham imagens em 3D (o que seria o ideal, segundo os autores). Na maioria das clínicas em todo o mundo nós medimos o volume dos NP com ressonâncias e imagens em 2D. Assim, o resultado deixa de nos apresentar uma medida objetiva do tamanho dos tumores.
- O estudo não separou os NP difusos dos NP nodulares, mas já sabemos que eles são muito diferentes na origem e no comportamento clínico (ver aqui mais informações sobre essa diferença importante). Assim, o estudo deixa de nos oferecer outra informação objetiva que poderia nos orientar na decisão de quando devemos tratar os NP.
- Outra fragilidade do estudo foi terem usado indicadores subjetivos (como dor e qualidade de vida) para avaliar o desfecho dos tratamentos, pois eles podem ser influenciados por fatores psicológicos, o chamado efeito placebo. Por exemplo, no estudo com o inibidor MEK que deu origem à liberação do quimioterápico selumetinibe nos Estados Unidos, a dor reduziu 2 pontos numa escala de 10 pontos nas crianças que usaram a droga, o que parece um efeito pequeno.
- O estudo não comparou a morbidade pós-operatória das crianças operadas com a morbidade das crianças que ficaram sem tratamento na mesma faixa etária, que parecia ser o objetivo inicial.
- Apesar de terem observado 4 pessoas (4,2%) que apresentaram transformação maligna dos seus NP, os autores não incluíram estes casos entre os 90 estudados como parte da evolução natural, pois consideraram a amostra pequena. Lamento, novamente, pois esta é uma das evoluções mais temidas dos NP e correspondem a 10% de todos os NP ao longo da vida.
- Finalmente, a conclusão do estudo me parece inadequada, pois é um pensamento circular, ou seja: usaram o momento no qual os médicos decidiram (no passado) tratar o NP (sem critérios científicos estabelecidos – que foi justamente a justificativa para eles realizarem o estudo) como critério para dizer que devemos tratar os NP o mais cedo possível.
Isto seria verdade se os autores tivessem encontrado melhor qualidade de vida nas crianças que foram tratadas mais cedo do que naquelas que foram tratadas mais tarde.
Ou tivessem comparado a qualidade de vida das crianças que foram operadas com a qualidade de vida das crianças que não foram operadas na mesma idade.
Mas eles não mostram esses dados.
Minha conclusão é que, infelizmente, ainda não temos uma regra geral para decidir o momento em que devemos tratar (com cirurgia ou medicamento) um neurofibroma plexiforme nas pessoas com NF1.
Portanto, continua valendo a decisão caso a caso, conforme nossa orientação geral para o tratamento dos NP disponível em nossa página: ver aqui.
Dr Lor