Um estudo científico bem conduzido na Austrália pelo grupo da Dra. Kathryn N. North avaliou o comportamento de crianças e adolescentes com neurofibromatose do tipo 1 (NF1), transtorno no espectro do autismo (TEA) e transtorno de déficit de atenção e hiperatividade (TDAH) e observou que estas 3 condições apresentam em comum o chamado transtorno do processamento sensorial (TPS) (ver aqui artigo completo em inglês).
Este texto recebeu a leitura e sugestões da Fernanda Siqueira (neuropsicopedagoga) e do Nilton Alves de Rezende (professor de medicina da UFMG).
Que ele seja útil à nossa comunidade NF!
Dr. Lor
Introdução
Para compreendermos bem o artigo australiano, precisamos lembrar como os estímulos (luz, som, calor, vibração etc) são percebidos pelos sensores do corpo (nervos nos olhos, orelhas, pele, músculos etc) para que sejam processados no cérebro de forma adequada para criar comportamentos que permitam a vida.
Como os estímulos são percebidos?
Para um estímulo qualquer ser percebido, ele precisa atingir um sensor (ou receptor) que seja sensível a ele. Por exemplo, a retina tem sensores para a luz, mas a pele não. Ao contrário, a pele tem sensores para o calor, mas a retina, não.
Quanto mais forte o estímulo, por exemplo, um som mais alto, mais ele ativa o sensor correspondente, no caso do som o sistema auditivo, que transmite um sinal para o cérebro, onde ele é processado (ver abaixo) para gerar uma resposta comportamental adequada àquele som.
Por outro lado, quanto mais sensível for um sensor, mais intensamente ele perceberá o estímulo. Por exemplo, um sistema auditivo mais sensível perceberá até os sons mais baixos e sutis.
A sensibilidade de cada sensor ao seu estímulo específico pode variar de acordo com a frequência com que o sensor está sendo estimulado. Quanto mais “novidade” for um estímulo, maior a sensibilidade do sensor. Quanto mais repetido for o estímulo, menor a sensibilidade do sensor.
A sensibilidade do sensor num certo momento é chamada de limiar de sensibilidade.
A medida do limiar da sensibilidade é a intensidade necessária para o estímulo ser percebido. Um limiar baixo significa que um pequeno estímulo ativa o sensor; um limiar alto significa que é preciso um estímulo mais forte para ativar o sensor.
Em resumo, quanto mais baixo o limiar, menor será o estímulo necessário para a criança sentir e reagir a ele (fenômeno chamado de sensibilização).
Quanto mais alto o limiar, maior será o estímulo necessário para a criança sentir e reagir a ele (fenômeno chamado de habituação).
Crianças com limiares sensoriais baixos são facilmente estimuladas por coisas que geralmente não incomodam as crianças neurotípicas. Por exemplo, crianças com NF1 podem ter maior sensibilidade tátil e por isso evitar andar descalças na grama ou areia ou expressar desconforto ou dor quando alguém escova seu cabelo.
Crianças com limiares sensoriais altos apresentam dificuldade para serem estimuladas por coisas que geralmente são percebidas pelas crianças neurotípicas. Por exemplo, crianças com NF1 podem demorar para perceber que estão chamando pelo seu nome ou não ter consciência do toque de outra pessoa, a menos que seja mais intenso.
O que é o processamento sensorial?
O processamento sensorial é a capacidade do sistema nervoso de detectar, integrar, modular e interpretar todas as informações captadas pelos diversos sensores que foram ativados pelos estímulos que nos chegam do ambiente e do nosso próprio corpo. As informações sensoriais processadas no sistema nervoso central, que permitem nossa adaptação ao ambiente, são:
- Visuais (luz, cores, sinais, símbolos, imagens, profundidade etc.)
- Auditivas (sons, fala, música, ruídos etc.)
- Espaciais (equilíbrio, orientação espacial, posição corporal, controle motor, expressão corporal etc.)
- Táteis (textura, formato, consistência, peso e volume de objetos etc.)
- Paladar (sabores, textura, consistência de alimentos e líquidos etc.)
- Olfato (odores, aroma de alimentos, perfumes etc.)
- Térmicas (percepção da temperatura, do calor e do frio, tolerância e aclimatação)
- Dolorosas (sensibilidade, tolerância, repercussões emocionais etc.)
- Internas fisiológicas (respiração, batimentos cardíacos, fadiga, sono, movimentos intestinais, volume da bexiga etc.)
- Internas psicológicas (humor, comportamento, identidade, sentimentos, desejo sexual etc.)
A integração de todas estas informações sensoriais é que nos permite reagir adequadamente às variações internas e do ambiente. Elas são utilizadas pelo sistema nervoso para que possamos perceber de forma contínua o nosso próprio corpo num determinado ambiente físico, assim como os indicadores e regras sociais para que nosso comportamento seja adequado a cada momento, em cada grupo de pessoas ou numa sociedade.
O que é o transtorno de processamento sensorial?
Quando as informações sensoriais são percebidas para mais ou para menos do que deveriam ser, a sua integração no sistema nervoso pode se tornar incorreta, gerando respostas comportamentais inadequadas, que são os Transtornos do Processamento Sensorial (TPS).
O estudo australiano mostrou que estes estímulos sensoriais podem ser percebidos em intensidades incorretas nas crianças com NF1, TDAH e TEA:
- Algumas percebem para menos os estímulos (ou seja, possuem limiar sensorial alto, por exemplo, não percebem quando chamam seu nome ou o sinal do despertador)
- Outras percebem para mais os estímulos (ou seja, possuem limiar sensorial baixo, por exemplo, um ruído habitual pode causar susto ou estresse)
A resposta comportamental aos estímulos sensoriais também pode variar nas as crianças com NF1, TDAH e TEA:
- Algumas reagem ativamente aos estímulos (por exemplo fugindo ou tapando os ouvidos etc.)
- Outras reagem passivamente aos estímulos (ignorando o estímulo ou reagindo internamente com ansiedade, por exemplo)
Cada criança com TPS apresenta comportamentos que misturam as características descritas acima. Por exemplo, uma criança pode ser muito sensível aos estímulos auditivos e reagir passivamente a eles com grande ansiedade, mas, ao mesmo tempo, ser pouco sensível aos estímulos táteis e reagir ativamente a eles procurando o contato com pessoas ou objetos frequentemente.
Ao contrário, outra criança pode ser pouco sensível aos estímulos auditivos e reagir ativamente buscando estímulos sonoros e fazendo barulho o tempo todo, mas, ao mesmo tempo, ser muito sensível aos estímulos táteis e reagir ativamente a eles evitando o contato com outras pessoas, certos tipos de roupas ou calçados.
O estudo australiano mediu o processamento sensorial auditivo, tátil, visual, oral, da posição corporal e do movimento nas crianças com NF1, TEA e TDAH e comparou com crianças neurotípicas. O resultado do estudo mostrou que o TPS foi observado em cerca de 60% das crianças com NF1, TEA e TDAH.
Sensibilidade aos estímulos e resposta comportamental
Podemos incorporar as definições anteriores num quadro que combina o nível de sensibilidade para determinado estímulo (para mais ou para menos) com a reação comportamental da criança (ativa ou passiva).
É o chamado Modelo de Quatro Quadrantes de Processamento Sensorial de Dunn. Este modelo, que se baseia em dados comportamentais e neurocientíficos, propõe que as crianças têm limiares neurais únicos para responder a informações sensoriais, que, por sua vez, afetam a forma como elas respondem ao seu ambiente cotidiano.
Respostas ativas ou passivas
A resposta de uma criança a um estímulo sensorial pode ser por meio de um comportamento ativo – agindo para aumentar ou reduzir o estímulo, – ou por meio de um comportamento passivo – ignorando o estímulo ou reagindo apenas internamente.
Combinando os limiares altos e baixos com as respostas ativas e passivas, temos o Modelo de Quatro Quadrantes de Processamento Sensorial de Dunn (abaixo).
|
Baixa sensibilidade
(limiar alto) |
Alta sensibilidade
(limiar baixo) |
| Reação ativa
tenta controlar o estímulo |
A |
B |
| Reação passiva
ignora o estímulo ou reage internamente |
C |
D |
Exemplos de uso do quadro (aqui apenas com estímulos auditivos, mas poderiam ser quaisquer outras modalidades de estímulos sensoriais):
No tipo A, seria uma criança que percebe pouco os estímulos sonoros e reage ativamente aumentando o som ou produzindo barulhos.
No tipo B, seria uma criança que percebe muito os estímulos sonoros e reage ativamente com redução do som, fuga, aversão ou tampando as orelhas.
No tipo C, seria uma criança que percebe pouco os estímulos sonoros e reage passivamente, sem resposta.
No tipo D, seria uma criança que percebe muito os estímulos sonoros e reage passivamente com manifestações internas de ansiedade e mal-estar.
Uma mesma criança com NF1 pode ser, por exemplo, do tipo A para estímulos sonoros, mas ser do tipo C para estímulos visuais.
Assim, o transtorno no processamento sensorial pode afetar significativamente a forma como uma criança responde e funciona em seu ambiente, resultando em comportamentos desafiadores, ansiedade, poucas habilidades adaptativas, poucas interações sociais, atraso no desenvolvimento da linguagem, redução no desempenho motor e acadêmico.
Esse impacto é exemplificado em indivíduos com TEA, que apresentam respostas atípicas a estímulos sensoriais em mais de 70% das crianças. As diferenças de processamento em crianças com TEA são relatadas em todas as modalidades sensoriais, incluindo domínios táteis, visuais e auditivos, com hipo e hiper-responsividade relatadas. Essas diferenças podem ter um impacto considerável nas interações sociais e no funcionamento do dia-a-dia. A hipersensibilidade ou hipossensibilidade a estímulos sensoriais é considerada um sintoma central do autismo, conforme conceituado pelo Manual Diagnóstico e Estatístico de Transtornos Mentais 5ª Edição – Revisão de Texto (DSM-5-TR), com alguns estudos sugerindo dificuldades de processamento sensorial, como hiper-responsividade ao som ou toque, podem estar entre os primeiros indicadores de TEA.
O estudo australiano
Depois da introdução acima, podemos retomar o estudo realizado pela equipe da Dra. Kathrin North. Apesar de haver outras modalidades de informações sensoriais, o estudo avaliou especificamente as auditivas, visuais, táteis, relacionadas ao movimento, posição corporal e orais (tato, paladar e olfato). Traduzimos e adaptamos algumas das partes do estudo, aquelas que nos pareceram mais úteis às pessoas leitoras sem formação biológica.
Em resumo, a equipe partiu da verificação que as dificuldades no processamento sensorial são freqüentemente encontradas em distúrbios do neurodesenvolvimento e podem afetar significativamente a forma como uma criança responde e funciona em seu ambiente. Porém, os estudos que examinam o processamento sensorial em crianças com neurofibromatose tipo 1 (NF1) são escassos. Então, o estudo examinou o processamento sensorial relatado pelos pais em uma amostra de 152 crianças com NF1.
Este estudo foi concebido para caracterizar o perfil de processamento sensorial de crianças com NF1. Abordamos três objetivos principais neste estudo:
(i) examinar a proporção de indivíduos com NF1 exibindo respostas incomuns a estímulos sensoriais em comparação com crianças com desenvolvimento típico (DT);
(ii) examinar o processamento sensorial, incluindo estilos e modalidades de resposta, de crianças com NF1 em comparação com um grupo controle de DT; e
(iii) examinar a associação entre características de processamento sensorial e idade, sexo e medidas dimensionais quantitativas de funcionamento intelectual, comportamentos autistas, sintomas de TDAH, sintomas internalizantes, habilidades sociais e funcionamento adaptativo.
Os pais/cuidadores de 152 crianças com NF1 e 96 crianças com desenvolvimento típico preencheram um questionário chamado Perfil Sensorial 2 (SP2), juntamente com questionários padronizados avaliando comportamentos autistas, sintomas de TDAH, sintomas internalizantes, funcionamento adaptativo e habilidades sociais. O funcionamento intelectual também foi avaliado.
O Perfil Sensorial 2 (SP2) [2] é um questionário de 86 itens para pais/cuidadores projetado para avaliar a função sensorial. Os itens são medidos em uma escala de 5 pontos que varia de “quase sempre” a “quase nunca”. O SP2 fornece um perfil sensorial de quatro quadrantes (de Dunn, ver acima): busca sensorial, hipersensibilidade, evitação sensorial e baixo registro.
Os dados do SP2 indicaram importantes problemas de processamento sensorial em crianças com NF1 em comparação com crianças com desenvolvimento típico. Mais de 40% das crianças com NF1 apresentaram diferenças no registro sensorial (falta de entrada sensorial) e foram incomumente sensíveis e evitativas a estímulos sensoriais.
Aproximadamente 61% das crianças com NF1 apresentaram diferenças na forma como respondem a estímulos sensoriais quando comparadas a um grupo controle com desenvolvimento típico (Figura 1).Essas dificuldades foram observadas igualmente em todas as idades e sexo e foram associadas a um maior grau de comportamentos autistas, sintomas de TDAH, habilidades adaptativas mais baixas, habilidades sociais mais pobres e aumento da ansiedade e sintomas afetivos.
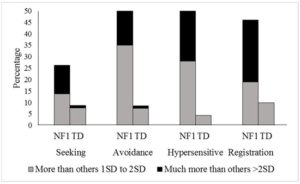
Figura 1. Porcentagem de NF1 e participantes de controle com desenvolvimento típico que excedem a faixa normal no SP2. Seeking busca ativa (Tipo A); Avoidance : evitação ativa (Tipo C); Hypersensitive: limiar baixo (Tipo D); Registration: limiar alto (Tipo B); Colunas cinza: transtornos mais presentes do que nas outras crianças; Colunas pretas: transtorno muito mais presente do que nas outras crianças.
Estes resultados destacam a importância de enfrentarmos as dificuldades de processamento multissensorial em casa e na escola ao decidirmos como apoiar uma criança com NF1 em todos os ambientes.
Discussão
Embora normalmente classificada como uma síndrome de predisposição tumoral, as complicações mais comuns em crianças com NF1 são dificuldades com o funcionamento social, comportamental, acadêmico e cognitivo. Aproximadamente 30-50% das crianças com NF1 atendem aos critérios para transtorno de déficit de atenção e hiperatividade (TDAH), até metade exibe comportamentos autistas e aproximadamente 25% atendem aos critérios diagnósticos para autismo, todos associados a dificuldades de processamento sensorial.
Apesar da alta prevalência dessas dificuldades de neurodesenvolvimento, os estudos que examinam o processamento sensorial em crianças com NF1 são escassos. As evidências recentes sugerem diferenças na forma como os bebês com NF1 processam informações auditivas, o que tem sido associado ao surgimento posterior de traços autistas. Alterações no processamento auditivo, incluindo a percepção das características temporais de um som, também foram detectadas em crianças com NF1, com algumas encontrando uma relação entre essas diferenças e funções, incluindo o grau de comprometimento da linguagem e distúrbio de comunicação e processamento fonológico.
Embora o estudo de sistemas sensoriais isolados (som ou visão ou tato etc.) dentro de uma condição genética forneça conhecimento valioso, a integração multissensorial é necessária para muitas funções, incluindo comunicação e linguagem.
Aumentar a conscientização não apenas sobre a gama de modalidades afetadas na NF1, mas também sobre o perfil sensorial e as respostas comportamentais relacionadas aos sentidos, será importante para informar e identificar intervenções sensoriais eficazes para crianças com NF1.
Este é o primeiro estudo a apresentar o perfil dos comportamentos em relação aos estímulos sensoriais em crianças com NF1 e sua associação com as características e funcionamento da criança.
Com exceção da modalidade visual, as crianças com NF1 apresentaram níveis mais altos de respostas incomuns a estímulos sensoriais em todas as dimensões de responsividade e modalidades sensoriais do que os controles DT.
Os tamanhos de efeito foram moderados a grandes. Essas dificuldades foram observadas igualmente em toda a faixa etária do estudo (3 a 15 anos) e em ambos os sexos. O risco relativo de desenvolver problemas de processamento sensorial foi de 3 a 9,6 vezes maior para crianças com NF1 em comparação com os controles DT, com hipersensibilidade considerada o maior risco, seguida por baixo registro, evitação sensorial e busca sensorial. Aproximadamente 61% das crianças com NF1 tiveram uma ou mais modalidades impactadas, com dificuldades comumente ocorrendo em várias modalidades e em várias áreas de responsividade.
Esses resultados sugerem que, se uma criança com NF1 tiver desafios de processamento sensorial, é provável que eles ocorram em várias áreas do processamento sensorial.
Eles podem ser sensoriais em busca de estímulos, ter baixo registro, evitar estímulos sensoriais e, ao mesmo tempo, ser hipersensíveis a estímulos sensoriais, pois esses padrões não são mutuamente exclusivos Por exemplo, uma criança com NF1 pode buscar entrada tátil brincando com materiais texturizados, mas, ao mesmo tempo, ter dificuldade em registrar certos sons. A mesma criança também pode evitar texturas específicas, como certas roupas, e ser sensível a ruídos altos.
Ao considerar como apoiar efetivamente as crianças com NF1, médicos, pais/cuidadores e educadores devem levar em consideração o processamento multissensorial e observar que o padrão de processamento sensorial de cada criança é único e pode variar em diferentes situações.
Observamos uma relação moderada a forte entre os escores SP2 e comportamentos autistas e sintomas de TDAH no estudo atual. Isso demonstra que crianças com NF1 que apresentam dificuldades de processamento sensorial também são mais propensas a exibir comportamentos autistas e sintomas de TDAH e sugere que as dificuldades de processamento sensorial são uma parte importante do fenótipo de neurodesenvolvimento da NF1.
Os resultados também demonstram um perfil de processamento sensorial semelhante na NF1 aos relatados em crianças com TDAH e TEA. Ou seja, todos os três grupos apresentam dificuldades com evitação sensorial, sensibilidade, registro e busca.
Embora a magnitude das dificuldades de registro tenha sido semelhante entre os três grupos, houve graus ligeiramente diferentes de gravidade entre os grupos para sensibilidade e evitação. Essas semelhanças nos perfis sensoriais podem fornecer informações sobre mecanismos subjacentes compartilhados.
O estilo sensorial de registro, para o qual uma magnitude semelhante de dificuldades de registro é experimentada em crianças com NF1, TEA e TDAH, reflete o grau em que as crianças se orientam ou “sintonizam” os estímulos ambientais. Uma criança com baixo registro pode apresentar falta de resposta ao seu nome ser chamado ou pode não responder ou se orientar adequadamente para estímulos sociais (rostos). Em teoria, uma criança pequena que não responde abertamente a novos estímulos sensoriais e sociais perde oportunidades de aprendizagem que são fundamentais para o desenvolvimento da comunicação social e habilidades adaptativas.
Uma explicação potencial para as dificuldades de registro (ou seja, hiporresponsividade) em crianças com NF1, que também foi sugerida para crianças com TDAH e TEA, pode estar relacionada a mecanismos de atenção alterados. A orientação comportamental é uma medida da capacidade de resposta de uma criança a novas informações sensoriais e teoriza-se que seja impulsionada pela interação com redes de atenção frontoparietal dorsal “de cima para baixo” e frontoparietal ventral “de baixo para cima”.
Supõe-se que essas redes de atenção sejam diferentes em indivíduos com autismo, potencialmente levando a uma tendência reduzida de orientar e atender a estímulos sociais. A evidência de um estudo de ressonância magnética funcional (fMRI) baseado em tarefas em crianças com NF1 indica diferenças semelhantes nas redes neurais associadas à orientação para estímulos sensoriais, sugerindo que anormalidades nessas redes neurais podem levar a uma orientação atencional ineficiente ou defeituosa da informação sensorial.
A fMRI em estado de repouso também pode ser uma ferramenta valiosa no exame dessas redes neurais e como elas se relacionam com o comportamento compartilhado entre os grupos de diagnóstico. As evidências indicam que cada domínio sensorial (registro, busca, sensibilidade e evitação) está associado a um padrão distinto de conectividade funcional intrínseca do cérebro. O baixo registro, por exemplo, tem sido associado a diferenças na conectividade nas redes frontoparietal e visual em crianças com TEA e TDAH. Embora diferenças na conectividade funcional tenham sido observadas em crianças com NF1, estudos futuros que examinam as relações entre respostas sensoriais e padrões de conectividade entre grupos (NF1, TEA e TDAH) podem nos ajudar a entender a sobreposição de sintomas sensoriais – circuitos neurais e relações de sintomas de TEA e TDAH entre os diagnósticos.
Alinhados com o estudo australiano, os estudos eletrofisiológicos da atividade cerebral humana indicaram que diferenças sensoriais na NF1 provavelmente ocorrerão em vários sistemas sensoriais. Dentro da visão, potenciais evocados visuais anormais e respostas de eletroencefalografia (EEG) foram relatados, com achados sugestivos de diferenças relacionadas à NF1 nos estágios posteriores do processamento visual e amplitude aumentada de oscilações alfa apoiando déficits no processamento sensorial básico em NF1. Recentemente, Begum-Ali e colaboradores (2021) usaram EEG para medir as respostas evocadas auditivas em bebês com NF1. Em relação aos controles, os lactentes com NF1 demonstraram uma latência prolongada ao apresentar uma resposta neural diferenciada ao detectar alterações nos estímulos auditivos. Isso sugere uma resposta atípica aos estímulos auditivos muito precocemente no desenvolvimento de crianças com NF1.
Modelos laboratoriais com animais com NF1 também fornecem uma oportunidade para estudar os mecanismos celulares e moleculares subjacentes às dificuldades sensoriais nas crianças com NF1. Os dados comportamentais e fisiológicos recentes em moscas drosófilas revelaram que a perda de Nf1 em neurônios sensoriais periféricos leva a erros de processamento sensorial. É importante ressaltar que esses erros contribuem para a detecção e / ou processamento prejudicado das pistas sociais, levando a déficits sociais na mosca com NF1. Isso sugere que um fluxo interrompido de informações sensoriais pode contribuir para efeitos no comportamento na NF1.
Apoiando essa ligação entre o gene NF1 e o processamento sensorial, Dyson e colaboradores (2022) mostraram que a perda de Nf1 em mosca drosófila resultou em hipersensibilidade tátil, que foi associada a déficits de transmissão sináptica dependentes de Ras indicativos de hiperexcitabilidade neuronal. Será importante para pesquisas futuras desenvolver resultados de processamento sensorial relevantes para a tradução para a clínica, que unam estudos em humanos e animais, para ajudar a entender melhor as vias causais dessas manifestações clínicas.
Os achados deste estudo têm implicações importantes para o manejo de crianças com NF1 e para o desenvolvimento de intervenções que visem apoiar seu funcionamento global. Embora o exame de transtornos de ansiedade e humor em crianças com NF1 tenha sido relativamente negligenciado, diversos estudos relatam aumento da ansiedade e dos sintomas depressivos em comparação com os controles de DT. Embora esses sintomas sejam considerados multifatoriais na etiologia, nossos resultados sugerem uma relação entre dificuldades de processamento sensorial e ansiedade e sintomas afetivos, indicando que eles podem ocorrer de forma associada em crianças com NF1. As dificuldades de processamento sensorial podem ter um papel na compreensão da prevalência aumentada de ansiedade e sintomas afetivos relatados na NF1 e destacam a importância de apoiar uma criança que experimenta diferenças de processamento sensorial.
Houve também várias relações moderadas a fortes observadas entre os domínios sensoriais e o comportamento funcional, incluindo habilidades adaptativas e habilidades sociais no estudo atual. Esses achados apóiam a literatura que relata uma estreita associação de dificuldades de processamento sensorial com limitações funcionais na vida diária e destacam ainda mais a importância de tratarmos as dificuldades de processamento sensorial na escola e em casa.
Educadores e profissionais da saúde devem levar em consideração o processamento multissensorial ao decidir como apoiar uma criança com NF1 em todos os ambientes. Os pais de crianças com NF1 e que apresentam dificuldades de processamento sensorial devem receber informações e recursos sobre comportamentos de processamento sensorial para garantir que estratégias de apoio sejam implementadas em todos os contextos.
As intervenções de processamento sensorial geralmente envolvem a criança em estratégias projetadas para treinar novamente os sentidos, incluindo auditivo, visual, tátil, proprioceptivo, oral, olfativo, vestibular e interoceptivo (o sentido envolvido na detecção de regulação interna, como frequência cardíaca e respiração).
Ainda precisamos esclarecer as incertezas sobre o resultado das intervenções sensoriais nas crianças com NF1. A evidência da eficácia dessas intervenções em outras condições de desenvolvimento, como o TEA, ainda é um trabalho em andamento, com a evidência geral limitada devido à falta de estudos de intervenções em larga escala.
Alguns efeitos positivos, no entanto, foram publicados apoiando a Intervenção de Integração Sensorial Ayres como uma intervenção baseada em evidências para problemas sensoriais no autismo. Esta intervenção individualizada baseada em jogos lúdicos utiliza atividades sensório-motoras que abordam dificuldades específicas identificadas em uma avaliação, que estão ligadas ao funcionamento da vida. Pesquisas futuras envolvendo ensaios clínicos em larga escala dentro de uma estrutura baseada em evidências são necessárias para determinar a eficácia dessa abordagem em geral, bem como em crianças com NF1.
Este estudo, como todo estudo científico, possui limitações. Embora o relatório dos pais ofereça uma avaliação ecologicamente válida do processamento sensorial na vida diária, ele se baseia apenas nas observações dos pais, e não nas experiências subjetivas da criança. A coleta de dados de vários informantes, incluindo a criança, pode fornecer uma melhor compreensão das diferenças de processamento sensorial em crianças com NF1. Estudos futuros que combinam paradigmas de processamento sensorial direcionados à criança (por exemplo, processamento auditivo de baixo nível ou EEG) e avaliação comportamental (SP2) podem fornecer informações sobre como as anormalidades nos sistemas sensoriais podem estar relacionadas aos perfis comportamentais do processamento sensorial.
Conclusões
Em resumo, os resultados deste estudo indicam que crianças com NF1 têm até nove vezes mais chances de experimentar diferenças no processamento sensorial do que seus pares não afetados. Além da visão, todas as modalidades sensoriais são afetadas, e o perfil de processamento sensorial da NF1 é amplamente semelhante ao de crianças com autismo e TDAH, mostrando evitação, sensibilidade, registro e dificuldades de busca, embora em diferentes graus de gravidade.
Dificuldades de processamento sensorial em crianças com NF1 podem levar a alterações comportamentais, como mau contato visual, evitação de locais ruidosos, ansiedade e reciprocidade social prejudicada, o que pode impactar no funcionamento diário.
Os modelos experimentais de animais com NF1 também apresentam deficiências no processamento sensorial que estão ligadas a déficits sociais, o que pode nos ajudar a entender os mecanismos de hiper/hiposensibilidade sensorial.
Como os déficits sensoriais são relativamente os mais tratáveis dos mecanismos do circuito e aparecem no início do desenvolvimento da NF1, o domínio sensorial é promissor para revelar mecanismos que podem contribuir para o desenvolvimento de dificuldades comportamentais de nível superior na NF1, como comprometimento social, mas também pode fornecer uma plataforma translacional não apenas para desenvolver biomarcadores, mas também para facilitar a busca contínua por novas abordagens terapêuticas na NF1.
Referências
- Dunn, W. The impact of sensory processing abilities on the daily lives of young children and their families: A conceptual model.
Infants Young-Child. 1997, 9, 23–35. [CrossRef]
- Dunn, W. The Sensory Profile 2 User’s Manual: Psychological Corporation; Psych Corporation: San Antonio, TX, USA, 2014.
- Baranek, G.T.; David, F.J.; Poe, M.D.; Stone, W.L.; Watson, L.R. Sensory experiences questionnaire: Discriminating sensory
features in young children with autism, developmental delays, and typical development. J. Child Psychol. Psychiatry 2006, 47,
591–601. [CrossRef]
- Buyuktaskin, D.; Iseri, E.; Guney, E.; Gunendi, Z.; Cengiz, B. Somatosensory Temporal Discrimination in Autism Spectrum
Disorder. Autism Res. 2021, 14, 656–667. [CrossRef] [PubMed]
- Hilton, C.L.; Harper, J.D.; Kueker, R.H.; Lang, A.R.; Abbacchi, A.M.; Todorov, A.; LaVesser, P.D. Sensory Responsiveness as a
Predictor of Social Severity in Children with High Functioning Autism Spectrum Disorders. J. Autism Dev. Disord. 2010, 40,
937–945. [CrossRef] [PubMed]
- Robertson, A.E.; Simmons, D.R. The Relationship between Sensory Sensitivity and Autistic Traits in the General Population.
J. Autism Dev. Disord. 2013, 43, 775–784. [CrossRef]
- Watson, L.R.; Patten, E.; Baranek, G.T.; Poe, M.; Boyd, B.A.; Freuler, A.; Lorenzi, J. Differential Associations Between Sensory
Response Patterns and Language, Social, and Communication Measures in Children With Autism or Other Developmental
Disabilities. J. Speech, Lang. Hear. Res. 2011, 54, 1562–1576. [CrossRef]
- Lane, A.E.; Young, R.L.; Baker, A.E.Z.; Angley, M.T. Sensory Processing Subtypes in Autism: Association with Adaptive Behavior.
J. Autism Dev. Disord. 2010, 40, 112–122. [CrossRef]
- Dunn, W.; Little, L.; Dean, E.; Robertson, S.S.; Evans, B. The state of the science on sensory factors and their impact on daily life
for children: A scoping review. Occup. Particip. Health 2016, 36, 3s–26s.
- Ahn, R.; Miller, L.; Milberger, S.; McIntosh, D. Prevalence of parents’ perceptions of sensory processing disorders among
kinder-garten children. Am. J. Occup. Ther. 2004, 58, 287–293. [CrossRef]
- Mallory, C.; Keehn, B. Implications of Sensory Processing and Attentional Differences Associated With Autism in Academic
Settings: An Integrative Review. Front. Psychiatry 2021, 12, 695825. [CrossRef]
Cancers 2023, 15, 3612
12. Baranek, G.; Boyd, B.; Poe, D.; Stone, W.L.; Watson, L.R. Hyperresponsive sensory patterns in young children with autism,
devel-opmental delay, and typical development. Am. J. Ment. Retard. 2007, 112, 233–245. [CrossRef]
13. Tomcheck, S.D.; Dunn, W. Sensory processing in children with and without autism: A comparative study using the short sensory
profile. Am. J. Occup. Ther. 2007, 61, 190–200. [CrossRef] [PubMed]
14. Elsabbagh, M.; Fernandes, J.; Webb, S.J.; Dawson, G.; Charman, T.; Johnson, M.H. Disengagement of Visual Attention in Infancy
is Associated with Emerging Autism in Toddlerhood. Biol. Psychiatry 2013, 74, 189–194. [CrossRef] [PubMed]
15. American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association
Publishing: Washington, DC, USA, 2022. [CrossRef]
16. Hadders-Algra, M. Emerging signs of autism spectrum disorder in infancy: Putative neural substrate. Dev. Med. Child Neurol.
2022, 64, 1344–1350. [CrossRef]
17. Sacrey, L.-A.R.; Zwaigenbaum, L.; Bryson, S.; Brian, J.; Smith, I.M.; Roberts, W.; Szatmari, P.; Roncadin, C.; Garon, N.; Novak, C.;
et al. Can Parents’ Concerns Predict Autism Spectrum Disorder? A Prospective Study of High-Risk Siblings From 6 to 36 Months
of Age. J. Am. Acad. Child Adolesc. Psychiatry 2015, 54, 470–478. [CrossRef]
18. Gutmann,D.; Ferner, R.E.; Listernick, R.; Korf, B.R.; Wolters, P.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers
2017, 3, 17004. [CrossRef] [PubMed]
19. Hyman,S.L.; Shores, A.; North, K.N. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1.
Neurology 2005, 65, 1037–1044. [CrossRef] [PubMed]
20. Lehtonen, A.; Howie, E.; Trump, D.; Huson, S.M. Behaviour in children with neurofibromatosis type 1: Cognition, executive
function, attention, emotion, and social competence. Dev. Med. Child Neurol. 2013, 55, 111–125. [CrossRef]
21. Pride, N.A.; North, K.N. The cognitive profile of NF1 children: Therapeutic implications. In Neurofibromatosis Type 1: Molecular
and Cellular Biology; Upadhyaya, M., Cooper, D.N., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 55–70.
22. Payne, J.M.; Hearps, S.J.C.; Walsh, K.; Paltin, I.; Barton, B.; Ullrich, N.; Haebich, K.M.; Coghill, D.; Gioia, G.A.; Cantor, A.; et al.
Reproducibility of cognitive endpoints in clinical trials: Lessons from neurofibromatosis type 1. Ann. Clin. Transl. Neurol. 2019, 6,
2555–2565. [CrossRef]
23. Payne, J.M.; Walsh, K.S.; A Pride, N.; Haebich, K.M.; Maier, A.; Chisholm, A.; Glad, D.M.; Casnar, C.L.; Rouel, M.; Lorenzo, J.;
et al. Social skills and autism spectrum disorder symptoms in children with neurofibromatosis type 1: Evidence for clinical trial
outcomes. Dev. Med. Child Neurol. 2020, 62, 813–819. [CrossRef]
24. Pride, N.A.; Payne, J.M.; North, K.N. The impact of adhd on the cognitive and academic functioning of children with nf1. Dev.
Neuropsychol. 2012, 37, 590–600. [CrossRef]
25. Mautner, V.-F.; Kluwe, L.; Thakker, S.D.; A Leark, R. Treatment of ADHD in neurofibromatosis type 1. Dev. Med. Child Neurol.
2002, 44, 164–170. [CrossRef] [PubMed]
26. Garg, S.; Lehtonen, A.; Huson, S.M.; Emsley, R.; Trump, D.; Evans, D.G.; Green, J. Autism and other psychiatric comorbidity in
neurofi-bromatosis type 1: Evidence from a population-based study. Dev. Med. Child Neurol. 2013, 55, 139–145. [CrossRef]
27. Morris, S.M.; Acosta, M.T.; Garg, S.; Green, J.; Legius, E.; North, K.; Payne, J.M.; A Weiss, L.; Constantino, J.N.; Gutmann, D.H.
Autism in neurofibromatosis type 1: Misuse of covariance to dismiss autistic trait burden. Dev. Med. Child Neurol. 2021, 63,
233–234. [CrossRef] [PubMed]
28. Chisholm, A.K.; Lami, F.; Haebich, K.M.; Ure, A.; Brignell, A.; Maloof, T.; Pride, N.A.; Walsh, K.S.; Maier, A.; Rouel, M.; et al.
Sex- and age-related differences in autistic behaviours in children with neurofibromatosis type 1. J. Autism Dev. Disord. 2023, 53,
2835–2850. [CrossRef] [PubMed]
29. Garg, S.; Green, J.; Leadbitter, K.; Emsley, R.; Lehtonen, A.; Evans, D.G.; Huson, S.M. Neurofibromatosis type 1 and autism
spectrum disorder. Pediatrics 2013, 132, e1642–e1648. [CrossRef] [PubMed]
30. Chisholm, A.; Haebich, K.; Pride, N.A.; Walsh, K.; Lami, F.; Ure, A.; Maloof, T.; Brignell, A.; Rouel, M.; Granader, Y.; et al.
Delineating the autistic phenotype in children with neuro-fibromatosis type 1. Mol. Autism 2022, 13, 3. [CrossRef]
31. Dunn,W.;Bennett, D. Patterns of Sensory Processing in Children with Attention Deficit Hyperactivity Disorder. OTJR: Occup.
Particip. Health 2002, 22, 4–15. [CrossRef]
32. Mangeot, S.; Miller, L.J.; McIntosh, D.; McGrath-Clarke, J.; Simon, J.; Hagerman, R.; Goldson, E. Sensory modulation dysfunction
in children with attention-deficit-hyperactivity-disorder. Dev. Med. Child Neurol. 2001, 43, 399–406. [CrossRef]
33. Marco, E.J.; Hinkley, L.B.N.; Hill, S.S.; Nagarajan, S.S. Sensory processing in autism: A review of neurophysiologic findings.
Pediatr. Res. 2011, 69, 48R–54R. [CrossRef]
34. Begum-Ali, J.; Kolesnik-Taylor, A.; Quiroz, I.; Mason, L.; Garg, S.; Green, J.; Johnson, M.H.; Jones, E.J.H.; The STAARS and EDEN
Teams. Early differences in auditory processing relate to Autism spectrum disorder traits in infants with Neurofibromatosis Type
1. J. Neurodev. Disord. 2021, 13, 22. [PubMed]
35. Rance, G.; Zanin, J.; Maier, A.; Chisari, D.; Haebich, K.; North, K.; Dabscheck, G.; Seal, M.L.; Delatycki, M.B.; Payne, J.M. Auditory
dysfunction among individuals with neurofibro-matosis type 1. JAMA Netw. Open 2021, 4, e2136842. [CrossRef] [PubMed]
36. Batista, P.; Lemos, S.; Rodrigues, C.; de Rezende, N. Auditory temporal processing deficits and language disorders in patients
with neurofibromatosis type 1. J. Commun. Disorders 2014, 48, 18–26. [CrossRef]
37. Norrix, L.W.; Plante, E.; Vance, R.; Boliek, C.A. Auditory-visual integration for speech by children with and without specific
lan-guage impairment. J. Speech Lang. Hear. Res. 2007, 50, 1639–1651. [CrossRef] [PubMed]
Cancers 2023, 15, 3612
38. Marton, K. Imitation of body postures and hand movements in children with specific language impairment. J. Exp. Child Psychol.
2009, 102, 1–13. [CrossRef]
39. Haebich, K.M.; A Pride, N.; Walsh, K.S.; Chisholm, A.; Rouel, M.; Maier, A.; Anderson, V.; Barton, B.; Silk, T.; Korgaonkar, M.; et al.
Understanding autism spectrum disorder and social functioning in children with neurofibromatosis type 1: Protocol for a
cross-sectional multimodal study. BMJ Open 2019, 9, e030601. [CrossRef]
40. Neurofibromatosis Conference Statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988,
45, 575–578.
41. Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.;
Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus
recommendation. Anesthesia Analg. 2021, 23, 1506–1513. [CrossRef]
42. Wechsler, D. Wechsler Intelligence Scale for Children, Fifth Edition: Australian and New Zealand Standardised Edition (WISC-V A&NZ);
Pearson: Bloomington, MN, USA, 2016.
43. Wechsler, D. Wechsler Preschool and Primary Scale of Intelligence, 4th ed.; Pearson: San Antonio, TX, USA, 2012.
44. Constantino, J.; Gruber, C.P. Social Responsiveness Scale, 2nd ed.; Western Psychological Services: Torrance, CA, USA, 2012.
45. Conners, C.K. Conners’ ADHD/DSM-IV Scales: Parent Version; Multi-Health Systems Inc.: New York, NY, USA, 1999.
46. Conners, C.K. Conners 3rd Edition Parent Toronto; Multi-Health Systems Inc.: North Tonawanda, NY, USA, 2008.
47. Harrison, P.; Oakland, T. Adaptive Behavior Assessment System, 3rd ed.; (ABAS-3); Western Psychological Services: Chicago, IL,
USA, 2015.
48. Elliott, S.N.; Gresham, F.M. Social skills interventions for children. Behav. Modif. 1993, 17, 287–313. [CrossRef]
49. Achenbach, T.M.; Rescorla, L.A. Manual for the ASEBA School-Age Forms and Profiles; University of Vermont, Research Center for
Children, Youth and Families: Burlington, VT, USA, 2001.
50. Little, L.; Dean, E.; Tomcheck, S.D.; Dunn, W. Sensory processing patterns in autism, attention deficit hyperactivity disorder, and
typical development. Phys. Occup. Ther. Pediatr. 2018, 38, 243–254. [CrossRef]
51. Ermer, J.; Dunn, W. The sensory profile: A discriminant analysis of children with and without disabilities. Am. J. Occup. Ther.
1998, 52, 283–290. [CrossRef] [PubMed]
52. Miller, L.J.; Anzalone, M.E.; Lane, S.J.; Cermak, S.A.; Osten, E.T. Concept evolution in sensory integration: A proposed nosology
for diagnosis. Am. J. Occup. Ther. 2007, 61, 135–140. [CrossRef]
53. Cheung, P.; Siu, A.M. A comparison of patterns of sensory processing in children with and without development disabilities. Res.
Dev. Disabil. 2009, 30, 1468–1480. [CrossRef]
54. Baranek, G.T.; Watson, L.R.; Boyd, B.A.; Poe, M.D.; David, F.J.; McGuire, L. Hyporesponsiveness to social and nonsocial sensory
stimuli in children with autism, children with developmental delays, and typically developing children. Dev. Psychopathol. 2013,
25, 307–320. [CrossRef] [PubMed]
55. Zwaigenbaum, L.; Bryson, S.E.; Rogers, T.; Toberts, W.; Brian, J.; Szatmari, P. Faculty opinions recommendation of behavioral
manifestations of autism in the first year of life. Int. J. Dev. Neurosci. 2005, 23, 143–152. [CrossRef] [PubMed]
56. Harris, N.S.; Courchesne, E.; Townsend, J.; A Carper, R.; Lord, C. Neuroanatomic contributions to slowed orienting of attention in
children with autism. Cogn. Brain Res. 1999, 8, 61–71. [CrossRef]
57. Vossel, S.; Geng, J.J.; Fink, G.R. Dorsal and ventral attention systems. Neuroscientist 2014, 20, 150–159. [CrossRef] [PubMed]
58. Mundy,P.; Newell, L. Attention, joint attention and social cognition. Curr. Dir. Psychol. Sci. 2007, 16, 269–274. [CrossRef]
59. Mundy,P.; Jarrold, W. Infant joint attention, neural networks and social cognition. Neural Netw. 2010, 23, 985–997. [CrossRef]
60. Pride, N.A.; Korgaonkar, M.S.; North, K.N.; Payne, J.M. Impaired engagement of the ventral attention system in neurofibromatosis
type 1. Brain Imaging Behav. 2018, 12, 499–508. [CrossRef]
61. Itahashi, T.; Fujino, J.; Sato, T.; Ohta, H.; Nakamura, M.; Kato, N.; Hashimoto, R.-I.; Di Martino, A.; Aoki, Y.Y. Neural correlates of
shared sensory symptoms in autism and attention-deficit/hyperactivity disorder. Brain Commun. 2020, 2, fcaa186. [CrossRef]
[PubMed]
62. Loitfelder, M.; Huijbregts, S.C.; Veer, I.M.; Swaab, H.; Van Buchem, V.; Schmidt, R.; Rombouts, S.A. Functional connectivity
changes and exec-utive social problems in neurofibromatosis type 1. Brain Connect. 2015, 5, 312–320. [CrossRef] [PubMed]
63. Ribeiro, M.J.; d’Almeida, O.C.; Ramos, F.; Saraiva, J.; Silva, E.D.; Castelo-Branco, M. Abnormal late visual responses and alpha
oscil-lations in neurofibromatosis type 1: A link to visual and attention deficits. J. Neurodev. Disord. 2014, 6, 4. [CrossRef]
64. Moscato, E.H.; Dubowy, C.; Walker, J.A.; Kayser, M.S. Social Behavioral Deficits with Loss of Neurofibromin Emerge from
Peripheral Chemosensory Neuron Dysfunction. Cell Rep. 2020, 32, 107856. [CrossRef]
65. Dyson, A.; Ryan, M.; Garg, S.; Evans, G.; Baines, R.A. Loss of NF1 in drosophila larvae causes tactile hypersensitivity and
impaired synaptic transmission at the neuromuscular junction. J. Neurosci. 2022, 42, 9450–9572. [CrossRef]
66. Walsh, K.S.; I Vélez, J.; Kardel, P.G.; Imas, D.M.; Muenke, M.; Packer, R.J.; Castellanos, F.X.; Acosta, M.T. Symptomatology of
autism spectrum disorder in a population with neurofibromatosis type 1. Dev. Med. Child Neurol. 2013, 55, 131–138. [CrossRef]
67. Johnson, N.S.; Saal, H.M.; Lovell, A.M.; Schorry, E.K. Social and emotional problems in children with neurofibromatosis type 1:
Evidence and proposed interventions. J. Pediatr. 1999, 134, 767–772. [CrossRef]
68. Sandbank, M.; Bottema-Beutel, K.; Crowley, S.; Cassidy, M.; Dunham, K.; Feldman, J.L. Project AIM: Autism intervention
me-ta-analysis for studies of young children. Psychol. Bull. 2020, 146, 1–29. [CrossRef]
Cancers 2023, 15, 3612
69. Camarata, S.; Miller, L.J.; Wallace, M.T. Evaluating sensory integration/sensory processing treatment: Issues and analysis. Front.
Integr. Neurosci. 2020, 14, 55. [CrossRef] [PubMed]
70. Pfeiffer, B.A.; Koenig, K.; Kinnealey, M.; Sheppard, M.; Henderson, L. Effectiveness of sensory integration interventions in
children with autism spectrum disorders: A pilot study. Am. J. Occup. Ther. 2011, 65, 76–85. [CrossRef]
71. Schaaf, R.C.; Benevides, T.; Mailoux, Z.; Faller, P.; Hunt, J.; van Hooydonk, E.; Freeman, R.; Leiby, B.; Sendecki, J.; Kelly, D. An
intervention for sensory difficulties in children with autism: A randomized trial. J. Autism Dev. Disord. 2014, 44, 1493–1506.