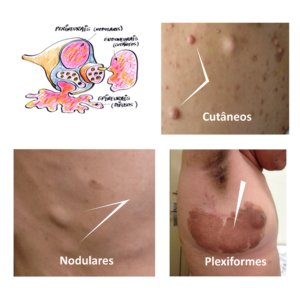
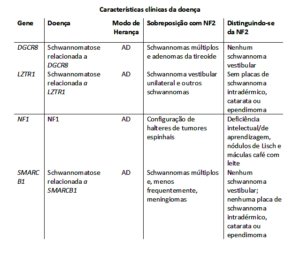
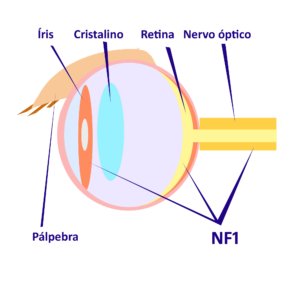
Damos continuidade à divulgação online dos capítulos escritos para a Edição Comemorativa dos 20 anos do CRNF, a ser lançada em novembro de 2024, apresentando hoje o texto excelente de dois nutricionistas, Dra. Aline e Dr. Márcio, que realizaram seus mestrados e doutorados envolvendo pessoas atendidas em nosso CRNF e contribuíram de forma importante e original internacionalmente para nossos conhecimentos sobre os problemas nutricionais nas pessoas com NF1. Agradecemos a sua colaboração neste tema tão interessante em nossa comunidade.
Dr. Lor
Aline Stangherlin Martins e Marcio Leandro Ribeiro de Souza
Nutricionistas e Doutores
no Programa de Pós Graduação
em Ciências Aplicadas à Saúde do Adulto
da Faculdade de Medicina da UFMG
As características nutricionais na NF1 começaram a ser estudadas recentemente e ainda são pouco conhecidas. Muitos fatores e características relacionadas à nutrição ainda precisam ser investigados nessa doença. A pesquisa de Souza e colaboradores, realizada no Centro de Referência em Neurofibromatoses de Minas Gerais, foi o primeiro estudo publicado que avaliou aspectos da alimentação em indivíduos com NF1, mais precisamente o consumo alimentar e a ingestão de nutrientes em indivíduos com a doença (SOUZA et al., 2015). Segundo os autores, 72% dos pacientes não atingiram suas recomendações de energia, especialmente os homens. Também foi observado uma ingestão inadequada de vitamina D, magnésio, cálcio e vitamina B6, tanto em homens quanto mulheres com a doença, e 100% dos participantes consumiram excesso de sódio. Os pacientes com NF1 não consumiram quantidades adequadas de fibras alimentares, vitamina A e vitamina C e foi observado um consumo excessivo de gordura saturada e lipídios. É importante observar que esse estudo foi apenas descritivo e não houve comparação com indivíduos sem a doença. Os autores discutem que o perfil de ingestão alimentar é ruim na NF1, assim como também é observado na população em geral (SOUZA et al., 2015).
Em uma comparação de ingestão de nutrientes entre indivíduos com NF1 e controles não acometidos pela doença, Souza e colaboradores observaram que o grupo NF1 apresentou um consumo menor de cálcio, ferro e vitamina A e um consumo maior de sódio, ácidos graxos poli-insaturados e ácido linoleico (ômega 6), quando comparado ao consumo do grupo controle. Não houve diferença entre os grupos para o consumo dos demais nutrientes. Nutrientes como cálcio, ferro, vitamina A, sódio e ômega 6 são relacionados normalmente com a saúde óssea, porém nesse estudo não houve correlação entre a ingestão de nutrientes e as características de baixa massa óssea e baixa densidade mineral óssea na NF1, observadas nesse estudo (SOUZA et al., 2020a).
Enquanto os estudos citados anteriormente avaliaram o consumo de nutrientes de forma isolada, Vilela e colaboradores avaliaram o padrão alimentar de adultos brasileiros com NF1. Nesse estudo, os autores observaram que 53,3% dos indivíduos com NF1 apresentaram um padrão alimentar ocidental, caracterizado por um consumo maior de alimentos não saudáveis, como margarina, maionese, sobremesas, frituras, embutidos, bebidas artificiais e alcoólicas. Nesse estudo, os indivíduos com NF1 e padrão alimentar ocidental apresentaram área muscular do braço menor que no grupo com padrão alimentar saudável, indicando que a alimentação ou dieta pode contribuir, mesmo em partes, para o fenótipo muscular comumente observado em indivíduos com NF1 (VILELA et al., 2020).
Enquanto estudos de avaliação da alimentação na NF1 são escassos, a maior parte dos estudos nessa doença apresentam parâmetros antropométricos como índice de massa corporal (IMC), peso, estatura e perímetro cefálico como características do estado nutricional nessa população, porém as prevalências e valores dessas variáveis não apresentam um consenso entre os estudos e, portanto, requerem uma investigação padronizada (SZUDEK; BIRCH; FRIEDMAN, 2000; TROVO-MARQUI et al., 2005; SOUZA et al., 2009; PETRAMALA et al., 2012).
A baixa estatura é mais frequente em indivíduos com NF1 quando comparados com pessoas sem a doença (MARTINS et al., 2016; MARTINS et al., 2018; SOUZA et al., 2019; SOUZA et al., 2020a). Souza e colaboradores encontraram 60% de baixa estatura (percentil menor que 5) e 54% de macrocrania (percentil maior que 95) em seu estudo com 183 pacientes com idades entre 1 e 67 anos comparada com curvas e tabelas para a população não acometida pela doença (SOUZA et al., 2009). Esses valores de baixa estatura e macrocrania diferem um pouco de outras pesquisas na NF1, todas estas realizadas com faixas etárias diferentes. Szudek e seus colaboradores encontraram 13% de baixa estatura e 24% de macrocrania em 569 crianças brancas americanas com NF1, o que resultou na elaboração de curvas de crescimento desses dois parâmetros para essa população (SZUDEK; BIRCH; FRIEDMAN, 2000). Trovó-Marqui e colaboradores também observaram baixa estatura em 40% dos seus pacientes com NF1, considerando valores de percentil menor que 3 e macrocrania em 51% dos indivíduos com a doença (percentil maior que 98) (TROVO-MARQUI et al., 2005). No estudo de Souza e colaboradores (2016), a prevalência de baixa estatura foi de 28,3%.
Na NF1, o IMC também apresenta valores diferentes quando se comparam os estudos. Petramala e colaboradores observaram IMC reduzido em 70 indivíduos com NF1 quando comparados a 40 voluntários sem a doença. As taxas de desnutrição, ou seja, indivíduos com IMC menor que 18,5 kg/m2, não foram descritas no estudo. Esse mesmo estudo mostrou apenas 5% de baixa estatura e 5% de macrocrania nos indivíduos avaliados (PETRAMALA et al., 2012). Rodrigues e colaboradores demonstraram estatura significativamente mais baixa para homens e mulheres com NF1 quando comparados a voluntários sem a doença. Esse mesmo estudo ainda encontrou diferença significativa com relação ao IMC, uma vez que o IMC de mulheres com NF1 foi maior que o IMC de controles saudáveis, contrariando o estudo de Petramala e colaboradores (2012) citado anteriormente, que encontrou IMC reduzido em indivíduos com NF1 comparados ao grupo controle (RODRIGUES et al., 2013).
O estudo de Koga e colaboradores buscou avaliar as características antropométricas (estatura e IMC) de 96 adultos japoneses com NF1 comparados a 288 voluntários não acometidos pela doença. A estatura foi menor na NF1 comparada aos controles. Quando estratificado por idade, as diferenças foram estatisticamente significativas para homens com idade entre 20 e 49 anos e mulheres de 30 a 39 anos. Por sua vez, o IMC dos homens com NF1 foi menor que o grupo controle. Não houve diferença entre as mulheres (KOGA et al., 2014). Essas mesmas características também foram confirmadas em outro estudo que buscava avaliar características metabólicas, nutricionais e musculares em japoneses com NF1 (KOGA; YOSHIDA; IMAFUKU, 2016). É interessante observar que existem divergências de resposta quando se analisa homens ou mulheres com NF1 para IMC e a justificativa para essa diferença precisa ser melhor investigada.
Martins e colaboradores encontraram menor peso e menor estatura em indivíduos com NF1 comparados com o grupo controle. Não houve diferenças no IMC, uma vez que os indivíduos foram pareados por essa variável (MARTINS et al., 2018). Outros estudos também não encontraram diferenças no IMC de indivíduos com NF1 quando comparados com pessoas sem a doença (SOUZA et al., 2019; SOUZA et al., 2020a). O estudo de Souza e colaboradores demonstrou que o baixo peso esteve presente em 10% dos voluntários avaliados e o excesso de peso em 31,7%. O IMC médio dos indivíduos com NF1 (23,86 ± 4,73 kg/m2) nesse estudo foi classificado como normal (eutrófico), da mesma forma que foi demonstrado no estudo de Petramala e colaboradores, com IMC médio de 22,5 ± 4,3 kg/m2, também em voluntários com NF1 (PETRAMALA et al., 2012; SOUZA et al., 2016).
De uma maneira geral, percebe-se que peso e estatura baixos são características clínicas comuns na NF1 com bastante variação nas prevalências entre os estudos, porém uma melhor investigação sobre os compartimentos corporais é necessária. Quanto à composição corporal na NF1, os estudos são ainda mais escassos e com resultados discrepantes. O estudo de Souza e colaboradores é um dos poucos estudos que abordaram especificamente a composição corporal na NF1, embora tenha usado métodos mais simples (dobras cutâneas e equações preditivas) para essa avaliação, e não utilizou um grupo controle, o que limita as conclusões. A área muscular do braço foi considerada baixa em 43,3% dos voluntários desse estudo, achado presente em 51,7% dos homens e 35,5% das mulheres. O percentual de gordura foi classificado como alto em 30% da amostra (SOUZA et al., 2016). Stevenson e colaboradores, em estudo realizado na Universidade de Utah, utilizaram a tomografia computadorizada periférica quantitativa para comparar os ossos e a musculatura esquelética de 40 crianças com NF1 e 380 voluntários não acometidos pela doença, com idades entre 5 e 18 anos. Este estudo demonstrou que crianças portadoras de NF1 apresentam menor área de secção transversal muscular que seus controles, porém sem maiores detalhamentos quanto à fisiopatologia deste achado (STEVENSON et al., 2005).
O método considerado padrão-ouro para avaliação da composição corporal é a absorciometria com raios-X de dupla energia (DXA). Estudos usando DXA para avaliação da massa magra ou composição corporal na NF1 não são encontrados facilmente. A maior parte dos estudos que utilizam a densitometria nessa doença avaliam apenas as características ósseas na doença, sem detalhamento dos compartimentos corporais. Dulai e colaboradores usaram a DXA em 23 crianças com NF1 com idades entre 5 e 17 anos, e o objetivo principal era avaliar a densidade mineral óssea que foi menor nestes voluntários. Os autores citam que a massa magra total ajustada pela altura foi normal, mas com uma diminuição do conteúdo mineral ósseo relativo à massa magra (DULAI et al., 2007). O estudo de Souza e colaboradores (2019) avaliou a composição corporal de adultos com NF1 através do DXA e não observou diferenças para percentual de gordura, massa gorda e massa magra quando comparados com indivíduos sem a doença. Porém a massa magra apendicular ajustada pelo IMC foi menor nos indivíduos com NF1. Esse estudo também demonstrou massa óssea e força muscular reduzidas em indivíduos com NF1, características comumente observadas nessa população (SOUZA et al., 2019).
Esses resultados divergentes entre os estudos reforçam a importância de uma investigação padronizada desses achados e de estudos que busquem entender as diferenças. Estudo em modelo animal encontrou os mesmos resultados e reforçou esses achados antropométricos e metabólicos observados nos indivíduos com NF1 (TRITZ et al., 2021).
O estudo de Souza e colaboradores demonstrou um gasto energético de repouso (GER) aumentado em indivíduos com NF1 após ajuste por peso ou massa magra ou massa magra apendicular (SOUZA et al., 2019). A hipótese inicial dos autores era que o GER fosse menor em indivíduos com NF1, uma vez que são indivíduos menores em tamanho (menor peso e estatura) e com menor massa muscular, características também demonstradas neste estudo, porém o GER aumentado foi observado na NF1 quando comparados com indivíduos sem a doença. O GER é influenciado por fatores como massa muscular, massa gorda, peso, idade, sexo, níveis de adipocinas como leptina e adiponectina, alterações metabólicas em processos patológicos, como hipo ou hipertireoidismo, entre outros (NIELSEN et al., 2000; JOHNSTONE et al., 2005; HALL et al., 2012; PSOTA; CHEN, 2013). Esse aumento de GER assemelha-se ao estudo de Leoni e colaboradores, realizado com a Síndrome de Costello, também uma Rasopatia, no qual os autores observaram um aumento de GER em indivíduos acometidos pela doença (LEONI et al., 2016). As causas para esse aumento de GER na NF1 precisam ser melhor investigadas e discutidas.
Pensando na assistência ao indivíduo com NF1 pelos profissionais de saúde, o estudo de Souza e colaboradores comparou 8 equações preditivas comumente usadas na prática clínica para estimar o GER com a calorimetria indireta (padrão-ouro) e observou que todas elas subestimam o GER em indivíduos com NF1 (SOUZA et al., 2020b). Em outro estudo, Souza e colaboradores fizeram uma comparação de equações que usam bioimpedância elétrica ou dobras cutâneas para avaliação da composição corporal com o método padrão-ouro DXA e observaram que as equações de bioimpedância elétrica apresentaram melhor adequação e acurácia, embora todas precisem ser usadas com cautela na NF1 (SOUZA et al., 2020c).
Alguns nutrientes são estudados de forma isolada na NF1. Um estudo demonstrou que indivíduos com NF1 apresentaram níveis séricos mais baixos de vitamina B12 quando comparados com indivíduos não acometidos pela doença (AYDIN; BUCAK, 2021). Outro nutriente comumente estudado na NF1 é a vitamina D. Alguns estudos observaram níveis reduzidos de vitamina nessa população e alguns estudos estudam discutem a importância dessa vitamina para o indivíduo com NF1, uma vez que pode atuar na saúde muscular e óssea, comumente afetados nos pacientes com essa doença (LAMERT et al., 2006; TUCKER et al., 2009; RICCARDI et al., 2020). Porém uma meta-análise recente não encontrou diferenças estatísticas nos níveis de vitamina D na NF1 e os autores sugerem que as alterações ósseas na NF1 podem ser resultantes de um mecanismo independente de vitamina D (KASPIRIS et al., 2024). Outro estudo demonstrou que os níveis de oito aminoácidos estavam alterados em indivíduos com NF1. Os autores demonstraram que arginina, glutamina, asparagina, serina e aspartato estavam aumentados nos indivíduos com a doença, enquanto cistina e prolina estavam reduzidos (OZ; KOYUNCU; GONEL, 2022).
Associado as características nutricionais já demonstradas, pesquisadores do CRNF observaram uma menor incidência de pacientes com diabetes Mellitus tipo 2 (DM) nos pacientes atendidos. Esse dado chamou a atenção dos profissionais, uma vez que o DM é uma doença altamente prevalente na população brasileira. Em revisão da literatura, verificou-se que dados apontavam para uma menor ocorrência de DM em pacientes com NF1 (ZAKA-UR-RAB; CHOPRA, 2005; OZHAN; OZGUVEN; ERSOY, 2013; MADUBATA et al., 2015).
Foram encontrados dois estudos apresentando relatos de caso envolvendo indivíduos com NF1, mas ambos relatavam que a associação entre DM1 e NF1 é pouco comum (ZAKA-UR-RAB; CHOPRA 2005; OZHAN; OZGUVEN; ERSOY, 2013). Um estudo desenvolvido a partir da análise de banco de dados, mostrou que pessoas com NF1 apresentaram uma chance significativamente menor de ter DM do que pessoas sem a doença (OR:0,4, IC95%: 0,3 – 0,4). Nesse estudo, foram incluídos pacientes com DM tipo1 e DM tipo 2 (MADUBATA et al., 2015).
Com objetivo de esclarecer sobre a menor frequência de diabetes em pacientes com NF1, Martins e colaboradores avaliaram a glicemia de jejum de 57 pacientes atendidos no CRNF. Os pacientes selecionados com NF1 foram pareados com controles não-NF1 selecionados do Estudo Longitudinal Brasileiro de Adultos Saúde de acordo com sexo, idade (35–74 anos) e índice de massa corporal (IMC) na proporção de 1:3. Em ambos os grupos, foram excluídos indivíduos com DM. O nível mediano de glicemia de jejum no grupo NF1 (86 mg/dl (intervalo, 56-127 mg/dl)) foi inferior ao do grupo de controle não-NF1 (102 mg/dl (intervalo, 85–146 mg/dl)) (P<0,001). A prevalência de glicemia de jejum >100 mg/dl no grupo NF1 (16%) foi menor do que no grupo controle não-NF1 (63%) (P<0,05). A chance de um alto nível de glicemia de jejum foi 89% menor no grupo NF1 (odds ratio, 0,112; IC 95%, 0,067–0,188) (P<0,05). Os autores concluíram que, adultos com NF1 apresentaram menores níveis de glicemia de jejum e menor prevalência de alto nível de glicemia de jejum em comparação com controles não-NF1 (MARTINS et al., 2016).
Diante dos dados que apontavam para menor frequência de DM e menor glicemia de jejum em pacientes com NF1, Martins e colaboradores desenvolveram uma pesquisa com objetivo de comparar a resistência à insulina (RI), que é um preditor para o desenvolvimento de diabetes, e alguns aspectos metabólicos que possuem relação com DM de pacientes com NF1 e indivíduos sem a doença. Foram avaliados 40 pacientes com NF1 pareados por sexo, idade e IMC a 40 controles da comunidade. Amostras de sangue foram coletadas para avaliação bioquímica. Avaliação do modelo de homeostase adiponectina (HOMA-AD), modelo de avaliação de homeostase resistência à insulina (HOMA-IR) e razão adiponectina/leptina (RAL) foram usados para identificar RI. Foram avaliados também os níveis de adipocitocinas leptina, visfatina, resistina e adiponectina. Os pesquisadores observaram que o valor HOMA-AD foi significativamente menor e a RAL significativamente maior no grupo NF1. Os níveis de glicemia de jejum, leptina e visfatina de pacientes com NF1 foram significativamente mais baixos e os níveis de adiponectina foram significativamente mais elevados do que os dos controles. Os autores concluíram que os níveis mais baixos de glicemia de jejum, leptina, visfatina e HOMA-AD, e níveis mais elevados de adiponectina e ALR podem estar relacionados ao aumento da sensibilidade à insulina e menor ocorrência de diabetes mellitus tipo 2 em pacientes com NF1 (MARTINS et al., 2018).
Em 2020, um estudo realizado por Kallionpää e colaboradores com 1410 pacientes, também observou menores taxas de DM2 em pacientes com NF1, corroborando as pesquisas realizadas no CRNF–MG (KALLIONPAA et al., 2021).
Estudos de intervenção nutricional na NF1 ou mesmo estudos que testaram a utilização de dieta ou nutrientes isolados na observação de características clínicas da doença são escassos. Como exemplo, pode-se citar o ômega 3. Mashour e colaboradores demonstraram in vitro que a utilização de ácidos graxos, principalmente ácido docosaexaenoico (DHA) e ácido araquidônico, pode representar possíveis substâncias reguladoras do desenvolvimento de TMBNP, resultantes da malignização de neurofibromas plexiformes (MASHOUR et al., 2005).
Um fenótipo comumente observado na NF1 é a força muscular reduzida, observado também no estudo de Souza e colaboradores (SOUZA et al., 2019). Segundo Sullivan e colaboradores, em um estudo com modelo animal, a perda da neurofibromina no músculo esquelético resulta em uma miopatia metabólica associada com acúmulo de gordura intramiocelular, aumento de triglicerídeos e aumento da atividade da ácido graxo sintase, e desregulação da função mitocondrial nesses músculos (SULLIVAN et al., 2014). Ainda pensando em prováveis mecanismos para explicar esse fenótipo de força muscular reduzida na NF1, Summers e colaboradores analisaram biópsias musculares de 6 crianças com NF1 e observaram fibrose e infiltração de células mononucleares, além de um considerável acúmulo de lipídio intramiocelular em todas as amostras, que apresentou correlação com redução da expressão da neurofibromina. A partir dessas observações, os autores testaram modelos animais que reproduziram todas as características patológicas observadas nas biópsias musculares humanas. Os animais apresentaram também acúmulo de lipídio intramiocelular que foi fortemente correlacionado com uma fraqueza muscular. Os autores, na sequência, testaram uma dieta enriquecida com ácidos graxos de cadeia média, reduzida em ácidos graxos de cadeia longa (AGCL) e aumentada em carnitina na dose de 300mg/kg/dia nesses modelos animais. Após 8 semanas, no grupo que recebeu AGCM e carnitina, houve um aumento de 45% na força muscular e redução de 71% no acúmulo de lipídio intramiocelular (SUMMERS et al., 2018). Essa associação da L-carnitina com ácidos graxos de cadeia média confirmou esses achados em um estudo clínico de fase 2 com crianças com NF1 e os autores sugerem que mais estudos em humanos testem essa estratégia para recuperação da força muscular (VASILJEVSKI et al., 2020; VASILJEVSKI et al., 2021). E um estudo em modelo animal observou que essa estratégia pode contribuir para melhorar a qualidade e porosidade óssea na NF1, o que precisa ser confirmado em humanos (O’DONOHUE et al., 2024).
Por fim, existem ainda estudos, por exemplo, associando constipação com a NF1, o que requer mais estudos (SRIDHAR et al., 2013; PEDERSEN et al., 2013; EJERSKOV et al., 2017). Outro estudo avaliou a presença de transtornos alimentares, como bulimia e anorexia, em indivíduos com NF1, uma vez que alterações dermatológicas da doença podem contribuir para distorções ou insatisfações com a imagem corporal (CHIAVARINO et al., 2023). Provavelmente no futuro novos achados e associações entre a doença e aspectos nutricionais podem aparecer e mais estudos se fazem necessários.
REFERÊNCIAS
AYDIN, H.; BUCAK, I.H. Neurofibromatosis type 1 and vitamin B12. Journal of the College of Physicians and Surgeons Pakistan, v. 31, n. 3, p. 353-355, 2021.
CHIAVARINO, F. et al. Eating disorders in Young patients with neurofibromatosis type 1. Journal of Paediatrics and Child Health, v. 59, n. 5, p. 723-728, 2023.
DULAI, S. et al. Decreased bone mineral density in neurofibromatosis type 1: results from a pediatric cohort. Journal of Pediatric Orthopedics, v. 27, n. 4, p. 472-475, 2007.
EJERSKOV, C. et al. Constipation in adults with neurofibromatosis type 1. Orphanet Journal of Rare Diseases, v. 12, n. 1, p. 139, 2017.
HALL, K.D. et al. Energy balance and its components: implications for body weight regulation. The American Journal of Clinical Nutrition, v. 95, n. 4, p. 989-994, 2012.
JOHNSTONE, A.M. et al. Factors influencing variation in basal metabolic rate include fat-free mass, fat mass, age, and circulating thyroxine but not sex, circulating leptin, or triiodothyronine. The American Journal of Clinical Nutrition, v. 82, n.5, p. 941-948, 2005.
KALLIONPÄÄ, R.A. et al. Haploinsufficiency of the NF1 gene is associated with protection against diabetes. Journal of Medical Genetics, v. 58, n. 6, p. 378-384, 2021.
KASPIRIS, A. et al. Bone mineral density, vitamin D and osseous metabolism indices in neurofibromatosis type 1: a systematic review and meta-analysis. Bone, v. 180, p. 116992, 2024.
KOGA, M. et al. Anthropometric characteristics and comorbidities in Japanese patients with neurofibromatosis type 1: a single institutional case-control study. The Journal of Dermatology, v. 41, n. 10, p. 885-889, 2014.
KOGA, M.; YOSHIDA, Y; IMAFUKU, S. Nutritional, muscular and metabolic characteristics in patients with neurofibromatosis type 1. The Journal of Dermatology, v. 43, n. 7, p. 799-803, 2016.
LAMMERT, M. et al. Vitamin D deficiency associated with number of neurofibromas in neurofibromatosis 1. Journal of Medical Genetics, v.43, n. 10, p. 810-813, 2006.
LEONI, C. et al. Understanding growth failure in Costello Syndrome: increased resting energy expenditure. The Journal of Pediatrics, v. 170, p. 322-24, 2016.
MADUBATA, C.C. et al. Neurofibromatosis type 1 and chronic neurological conditions in the United States: an administrative claims analysis. Genetics in Medicine, v. 17, n. 1, p. 36-42, 2015.
MARTINS A.S. et al. Lower fasting blood glucose in neurofibromatosis type 1. Endocrine Connections, v. 5, p. 28-33, 2016.
MARTINS A.S. et al. Increased insulin sensitivity in individuals with neurofibromatosis type 1. Archives of Endocrinology and Metabolism, v.62, n. 1, p. 41-46, 2018.
MASHOUR, G.A. et al. Differential modulation of malignant peripheral nerve sheath tumor growth by omega-3 and omega-6 fatty acids. Oncogene, v. 24, p. 2367-2374, 2005.
NIELSEN, S. et al. Body composition and resting energy expenditure in humans: role of fat, fat-free mass and extracelular fluid. International Journal of Obesity and Related Metabolic Disorders, v. 24, n. 9, p. 1153-1157, 2000.
O’DONOHUE, A.K. et al. Dietary intervention rescues a bone porosity phenotype in a murine model of neurofibromatosis type 1 (NF1). PLos One, v. 19, n. 6, p. e0304778, 2024.
OZ, O.; KOYUNCU, I.; GONEL, A. A pilot study for investigation of plasma amino acid profile in neurofibromatosis type 1 patients. Combinatory Chemistry & High Throughput Screening, v. 25, n. 1, p. 114-122, 2022.
OZHAN, B.; OZGUVEN, A. A.; ERSOY, B. Neurofibromatosis type 1 and diabetes mellitus: an unusual association. Case Reports in Endocrinology, v. 2013, p. 689107, 2013.
PEDERSEN, C.E. et al. Constipation in children with neurofibromatosis type 1. Journal of Pediatric Gastroenterology and Nutrition, v. 56, n. 2, p. 229-232, 2013.
PETRAMALA, L. et al. Bone mineral metabolism in patients with neurofibromatosis type 1. Archives of Dermatological Research, v. 304, n. 4, p. 325-331, 2012.
PSOTA, T.; CHEN, K.Y. Measuring energy expenditure in clinical populations: rewards and challenges. European Journal of Clinical Nutrition, v. 67, n. 5, p. 436-442, 2013.
RICCARDI, C. et al. Understanding the biological activities of vitamin D in type 1 neurofibromatosis: new insights into disease pathogenesis and therapeutic design. Cancers (Basel), v. 12, n. 10, p. 2965, 2020.
RODRIGUES, L.O. et al. Non-invasive endothelial function assessment in patients with neurofibromatosis type 1: a cross-sectional study. BMC Cardiovascular Disorders, v. 13, p. 18, 2013.
SOUZA, J.F. et al. Neurofibromatose tipo 1: mais comum e grave do que se imagina. Revista da Associação Médica Brasileira, v. 55, n. 4, p. 394-399, 2009.
SOUZA, M.L.R. et al. Body composition in adults with neurofibromatosis type 1. Revista da Associação Médica Brasileira, v. 62, n. 9, p. 831-836, 2016.
SOUZA, M.L.R et al. Nutrient intake in neurofibromatosis type 1: a cross-sectional study. Nutrition, v. 31, n. 6, p. 858-862, 2015.
SOUZA, M.L.R. et al. Increased resting metabolism in neurofibromatosys type 1. Clinical Nutrition ESPEN, v. 32, p. 44-49, 2019.
SOUZA, M.L.R. et al. Reduced bone mineral content and density in neurofibromatosis type 1 and its association with nutrient intake. Revista da Associação Médica Brasileira, v. 66, n. 5, p.666-672, 2020a.
SOUZA, M.L.R. et al. Resting energy expenditure in neurofibromatosis type 1 : indirect calorimetry versus predictive equations. Journal of Human & Clinical Genetics, v. 2, n. 1, p. 9-14, 2020b.
SOUZA, M.L.R. et al. Assessment of adiposity in neurofibromatosis type 1 : comparison between dual energy X-ray absorptiometry and conventional methods. Journal of Human & Clinical Genetics, v. 2, n. 1, p. 15-22, 2020c.
SRIDHAR, H. et al. Chronic Constipation Caused by Neurofibromatous Proliferation in A Case of Von Recklinghausen’s Disease – A Case Report. Journal of Clinical and Diagnostic Research, v. 7, n. 9, p. 2001-2003, 2013.
STEVENSON, D.A. et al. Case control study of the muscular compartments and osseous strength in neurofibromatosistype1 using peripheral quantitative computed tomography. Journal of Musculoskeletal & Neuronal Interactions, v. 5, n. 2, p. 145-149, 2005.
SULLIVAN, K. et al. NF1 is a critical regulator of muscle development and metabolism. Human Molecular Genetics, v. 23, n. 5, p. 1250-1259, 2014.
SUMMERS, M.A. et al. Dietary intervention rescues myopathy associated with neurofibromatosis type 1. Human Molecular Genetics, v. 27, n. 4, p. 577-588, 2018.
SZUDEK, J.; BIRCH, P.; FRIEDMAN, J.M. Growth in North American white children with neurofibromatosis 1 (NF1). Journal of Medical Genetics, v. 37, n. 12, p. 333-338, 2000.
TRITZ, R. et al. Nf1 heterozygous mice recapitulate the anthropometric and metabolic features of human neurofibromatosis type 1. Translational Research, v. 228, p. 52-63, 2021.
TROVÓ-MARQUI, A.B. et al. High frequencies of plexiform neurofibromas, mental retardation, learning difficulties, and scoliosis in Brazilian patients with neurofibromatosis type 1. Brazilian Journal of Medical and Biological Research, v. 38, n. 9, p. 144-147, 2005.
TUCKER, T. et al. Bone health and fracture rate in individuals with neurofibromatosis 1 (NF1). Journal of Medical Genetics, v. 46, n. 4, p.259-265, 2009.
VASILJEVSKI, E.R. et al. Evaluating modified diets and dietary supplement therapies for reducing muscle lipid accumulation and improving muscle function in neurofibromatosis type 1 (NF1). PLoS One, v. 15, n. 8, p. e0237097, 2020.
VASILJEVSKI, E.R. et al. L-carnitine supplementation for muscle weakness and fatigue in children with neurofibromatosis type 1: a phase 2a clinical trial. American Journal of Medical Genetics Part A, v. 185, n. 10, p. 2976-2985, 2021.
VILELA, D.L.S. et al. Dietary patterns of Brazilian adults with neurofibromatosis type 1. Revista Chilena de Nutrición, v. 47, n. 5, p. 772-781, 2020.
ZAKA-UR-RAB, Z.; CHOPRA, K. Diabetes mellitus in neurofibromatosis I: an unusual presentation. Indian Pediatrics, v. 42, n. 2, p. 185-186, 2005.