Sim, temos mais um motivo de esperança de que haverá – no futuro – algum medicamento eficiente para os neurofibromas, pois outra droga foi capaz de reduzir parcialmente os neurofibromas plexiformes em quase a metade das crianças e adultos com neurofibromatose do tipo 1 (NF1) que foram tratados com MIRDAMETINIBE (um inibidor de uma das vias – MEK – do metabolismo celular).
Apresentamos abaixo um resumo dos resultados do estudo que justificou a liberação da droga (ReNeu: A Pivotal, Phase IIb Trial of Mirdametinib in Adults and Children With Symptomatic Neurofibromatosis Type 1-Associated Plexiform Neurofibroma ver aqui o artigo completo – em inglês). e nossos comentários em seguida.
Atenção profissionais da saúde: amanhã postaremos a Avaliação Técnica e Científica do estudo que motivou a aprovação da droga Mirdametinibe para tratamento de neurofibromas plexiformes sintomáticos inoperáveis nos Estados Unidos, realizada pela Dra. Luíza de Oliveira Rodrigues a pedido do CRNF e da AMANF, destinada a fornecer informações científicas aos profissionais da saúde, ao Sistema Único de Saúde, aos órgãos públicos reguladores (ANVISA e CONITEC) e planos de Saúde Suplementar.
Os resultados do estudo clínico
Os autores do estudo pertencem a diversos centros de atendimento em NF1 nos Estados Unidos e reuniram informações sobre 58 adultos (de 18 a 69 anos) e 56 crianças (de 2 a 17 anos) com NF1 e com neurofibromas plexiformes (NP) sintomáticos que não podiam ser removidos cirurgicamente.
Os principais sintomas iniciais eram: dor (90% dos adultos e 70% das crianças) e deformidade estética (52% dos adultos e 50% das crianças).
Todas estas pessoas voluntárias receberam cápsulas ou comprimidos para suspensão oral de MIRDAMETINIBE (2 mg por metro quadrado de superfície corporal) duas vezes por dia, máximo de 4 mg duas vezes por dia), independentemente da ingestão de alimentos, em ciclos de 28 dias (sendo 3 semanas com a droga e 1 semana sem a droga).
O objetivo principal foi medir o volume do tumor antes e durante 25 ciclos de tratamento, para saber se a droga seria capaz de reduzir o tamanho do NP em 20% ou mais do volume inicial.
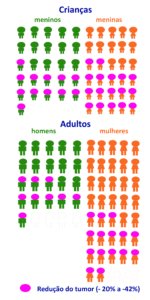
Este resultado foi observado em 24 dos 58 adultos (41%) e em 29 das 56 crianças (52%). Nos demais voluntários, o NP continuou estável ou interromperam o tratamento por efeitos colaterais ou crescimento dos tumores acima de 20% (ver figura acima com os resultados). .
Nos adultos e crianças que responderam positivamente ao MIRDAMETINIBE, a melhor resposta foi a redução de 41% nos NP dos adultos e redução de 42% dos NP nas crianças (mediana). Quer dizer, em 41% dos adultos os tumores reduziram 41% e em 52% das crianças os tumores reduziram 42% no seu volume.
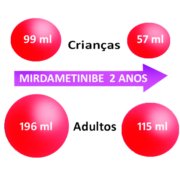
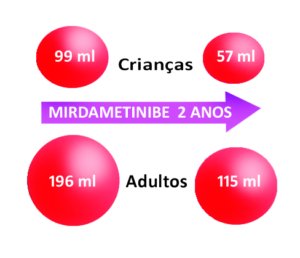
Nas crianças – que responderam ao tratamento, – o volume inicial dos tumores era de 99 ml (ver representação volumétrica na figura abaixo) e passou para 57 ml.
Nos adultos – que responderam ao tratamento, – o volume inicial dos tumores era de 196 ml e passou para 115 ml.
Veja abaixo uma figura representativa da mudança no volume dos tumores
Os autores não encontraram qualquer relação entre o efeito do MIRDAMETINIBE e o tamanho do tumor no início do tratamento. Além disso, a idade da pessoa também não influenciou a resposta à droga nos adultos e nem nas crianças.
Na escala de dor de 11 pontos, o uso do MIRDAMETINIBE reduziu a dor em 1,3 pontos nos adultos e 0,8 pontos nas crianças.
Os autores consideram seus resultados comparáveis aos resultados do estudo que serviu de base para a aprovação do SELUMETINIBE (ver estudo Sprint) tanto nos Estados Unidos quanto no Brasil.
Todas as crianças e adultos que usaram o MIRDAMETINIBE apresentaram um ou mais dos efeitos colaterais: acne (43 e 78%, respectivamente), diarreia (38 e 48%), náusea (21 e 36%), vômitos (14 e 28%), cansaço (9 e 21%), pele seca (14% e 14%), queda de cabelos (12 e 12%), redução da função cardíaca (20 e 12%), alteração da CPK sanguínea (20 e 10%), micose nas unhas (30 e 2%) e outros.
A conclusão dos autores é de que este foi o maior estudo (em número de pessoas) multicêntrico sobre NP em pessoas com NF1 e o MIRDAMETINIBE mostrou redução profunda e durável de volume do NP e melhora da dor e da qualidade de vida, sendo bem tolerado em adultos e crianças.
Comentários da equipe do CRNF
Lemos detalhadamente e discutimos este estudo e apresentamos alguns comentários a seguir.
Esta nova droga indica que inibir a via MEK do metabolismo celular pode ser um caminho promissor para a descoberta de medicamentos eficientes no futuro, mas o MIRDAMETINIBE ainda não é o medicamento ideal – com o qual nós e todas as pessoas com NF1 sonhamos – para melhorar o sofrimento das pessoas com NF1 e neurofibromas plexiformes (ver aqui mais informações sobre os neurofibromas plexiformes).
Por que o MIRDAMETINIBE ainda não é o medicamento ideal?
1 – Houve pouca redução no tamanho dos tumores
A melhor redução do volume dos NP foi de cerca de 40% em menos da metade das pessoas (adultos e crianças) que conseguiram completar o estudo por dois anos.
Nossa dúvida é se esta redução do tumor – nas pessoas em que ele ocorreu – traria realmente benefício para a qualidade de vida das pessoas com NF1?
2 – Em cerca de 20% das crianças e 5% dos adultos o NP continuou crescendo mesmo com a quimioterapia
Nestas pessoas a droga não foi capaz de agir nos NP, fazendo com que o tratamento fosse suspenso.
3 – Em cerca de 20% das crianças e 15% dos adultos o NP permaneceu do mesmo tamanho
Isto significa que somando estes tumores que permaneceram estáveis com aqueles que continuaram crescendo, em mais da metade das pessoas a droga não fez nenhum efeito nos tumores.
Isto indica que os fatores biológicos que promovem o crescimento dos NP são diferentes entre as pessoas e o MIRDAMETINIBE não foi capaz de inibir todos eles.
4 – Todas as pessoas apresentaram efeitos colaterais
Mesmo que os efeitos não tenham sido fatais, foram graves o suficiente para cerca de 20% das crianças e 40% dos adultos abandonarem o tratamento.
Assim, o uso clínico desta quimioterapia seria limitado pelos efeitos colaterais que a pessoa com NF1 seria capaz de tolerar em troca de uma redução parcial do seu NP.
5 – A redução da dor foi de cerca de 1 a 2 pontos numa escala de 0 a 10 pontos
Isto quer dizer que se a dor antes era de 5, por exemplo, ela passou para 3 no grupo que recebeu o MIRDAMETINIBE. Será que foi mesmo o MIRDANETINIBE o responsável pela diminuição da dor?”
6 – A redução do volume não apresentou relação com a melhora da dor
Não houve relação entre a redução no tamanho do tumor com a mudança da dor e da qualidade de vida. Portanto, apenas tentar reduzir o volume do NP não trouxe benefício obrigatoriamente para as pessoas que usaram o MIRDAMETINIBE.
Nossa interpretação, portanto, é que esta droga ainda não é o medicamento ideal, pois apresenta pouco benefício em relação aos seus riscos e o provável custo elevado, considerando-se as drogas semelhantes que já têm preço no mercado.
Outras dúvidas
Além disso, nossa análise do estudo nos trouxe algumas dúvidas.
1 – Qual foi a indicação para o tratamento dos NP nas pessoas que se tornaram voluntárias?
As razões pelas quais os NP não podiam ser removidos parcial ou completamente por meio de cirurgia não foram informadas pelos autores, nem nos anexos disponíveis na internet.
Por exemplo, o volume dos tumores variou de 1 ml a 3,4 litros nos adultos e de 5 ml a 3,6 litros nas crianças. É difícil imaginar uma situação clínica na qual um tumor periférico (como são todos os NP) de 1 a 5 ml não possa ser retirado cirurgicamente quando apresenta algum sintoma intratável.
2 – O crescimento ou não dos tumores antes do tratamento
Em 53% dos adultos e 62% das crianças os NP estavam em crescimento antes do uso do MIIRDAMETINIBE.
Isto quer dizer que nos demais voluntários os NP apresentavam estabilidade, portanto, a indicação de tratamento nestes casos se torna menos evidente, ou seja, não deveriam apenas ser observados clinicamente por mais tempo?
3 – Foi mesmo o MIRDAMETINIBE que reduziu 1 a 2 pontos na escala de dor?
Nos questionários sobre a qualidade de vida, cerca de 70% dos adultos e parentes das crianças relataram melhora com o uso da droga.
Isso indica que mais pessoas apresentaram melhora da qualidade de vida do que aquelas que tiveram redução no volume dos NP (cerca de 40 a 50%), ou seja, não haveria relação entre qualidade de vida e tamanho do tumor.
Em outras palavras, o simples fato de estarem “recebendo um medicamento” poderia reduzir a dor e melhorar a qualidade de vida, ou seja, seria o chamado efeito placebo?
5 – Por que não apresentaram as variantes genéticas encontradas nas pessoas voluntárias?
Teria sido uma contribuição muito importante deste estudo se tivessem incluído uma análise da variante patogênica como um possível determinante de quais pacientes respondem melhor ao tratamento, uma recomendação que foi debatida no Congresso do de 2024 no Arizona, e que seria outro ponto para direcionar melhor o uso de quaisquer novas drogas.
4 – Haveria interesses financeiros motivando os pesquisadores?
Algumas informações trazem alguma insegurança quanto a possíveis conflitos de interesse, pois muitos dos pesquisadores possuem vínculo com o laboratório fabricante do MIRDAMETINIBE.
O estudo que resumimos acima foi publicado na revista Journal of Clinical Oncology por 40 pesquisadores de várias instituições científicas norte-americanas, dos quais 13 deles têm relações comerciais com o Laboratório Spring-Works, fabricante da droga e financiador do estudo (ver aqui o artigo completo – em inglês).
Ficaríamos mais seguros sobre a isenção dos pesquisadores se TODOS eles não tivessem conflitos de interesse com a bilionária indústria de medicamentos.
A liberação rápida pela FDA
A agência norte-americana FDA (Food and Drug Administration) – semelhante à ANVISA no Brasil – liberou a droga MIRDAMETINIBE para ser usada no tratamento de neurofibromas plexiformes SINTOMÁTICOS E INOPERÁVEIS (ver aqui o comunicado) com base no estudo resumido acima e publicado em novembro de 2024 (ver aqui o artigo completo – em inglês).
O processo de liberação ocorreu de forma rápida (menos de 2 anos) porque o Laboratório inscreveu a droga na FDA como destinada a doença rara e doença órfã, o que facilita sua aprovação com menos exigências e comprovações de eficiência e segurança (ver ao final estudo Silvana e col.).
Um sinal de certa pressa na aprovação está na afirmação da FDA de que a droga MIRDAMETNIBE está aprovada para tratamento dos “neurofibromas plexiformes sintomáticos não passíveis de ressecção completa”.
Esta condição (ressecção completa) não faz parte do estudo que justificou a liberação e, além disso, sabemos que a imensa maioria dos neurofibromas não pode ser retirada cirurgicamente de forma completa (ver informações sobre os plexiformes).
Se aplicada rigorosamente esta recomendação da FDA ela tornaria o MIRDAMETINIBE indicado para praticamente TODOS os neurofibromas plexiformes.
Ou seja, ampliaria o mercado de venda do produto de cerca de 5% dos pacientes com NF1 (que, de fato, apresentam NP sintomáticos e inoperáveis) – segundo nossa avaliação – para 50% da população com NF1. Uma simples palavra que pode resultar em lucros significativos.
Conclusão da equipe do CRNF
A principal questão diante de uma pessoa com NF1 e NP é saber se, quando e por quais motivos, devemos ou não realizar um tratamento, seja cirúrgico ou medicamentoso (Ver aqui mais sobre esta questão).
Os resultados apresentados acima com o MIRDAMETINIBE parecem, – a nós e, inclusive, aos autores do estudo, – que são semelhantes aos encontrados com o SELUMETINIBE.
Portanto, nossa sugestão é que – se e – quando o MIRDANETINIBE estiver disponível no Brasil, o seu uso seja orientado pelos mesmos passos clínicos que apresentamos anteriormente em nossa página (ver aqui).
Aguarde:
Amanhã postaremos a Avaliação Técnica do estudo que motivou a aprovação da droga Mirdametinibe para tratamento de neurofibromas plexiformes sintomáticos inoperáveis nos Estados Unidos realizada pela Dra. Luíza de Oliveira Rodrigues a pedido do CRNF e da AMANF, destinada a fornecer informações a profissionais da saúde, ao Sistema Único de Saúde, aos órgãos públicos reguladores (ANVISA e CONITEC) e planos de Saúde Suplementar.