Pergunta da R.R. do Rio de Janeiro: “Meu filho tem 7 anos, tem NF1 e é o menor da turma e o endocrinologista pediu a dosagem do hormônio e veio 6 ng/mL. Então ele indicou hormônio do crescimento. Fora isso, meu menino está bem. Devemos usar?”
Cara R., obrigado pela sua pergunta, pois muitas famílias nos trazem dúvidas parecidas.
Iniciamos nossa resposta dizendo que não se deve tomar a decisão de iniciar o tratamento com Hormônio do Crescimento (GH) baseando-se apenas no valor de 6 ng/mL.
A indicação de tratamento da baixa estatura com GH é complexa e depende de uma avaliação clínica e endocrinológica completa, levando em conta os riscos e benefícios deste tratamento e considerando os valores e objetivos da família.
Comecemos pelo resultado do exame, o valor de GH de 6 ng/mL. Alguns valores de referência podem variar, com alguns laboratórios considerando a faixa normal para a idade entre 0,05 e 5,11 ng/mL.
Se for um valor isolado, sem estímulo, tem pouco valor diagnóstico isolado. A secreção de GH é pulsátil e varia ao longo do dia, portanto, um nível como este pode ser normal, baixo ou alto.
O diagnóstico de deficiência de GH não se faz com uma única dosagem, mas sim com testes de estímulo, onde se espera um pico maior que 10 ng/mL para descartar a deficiência. Um valor de 6 ng/mL, dependendo do contexto, pode ser considerado inseguro e necessita de investigação adicional.
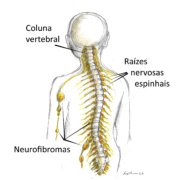
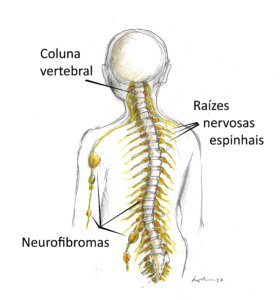
GH na NF1
A baixa estatura é comum (20-30% das pessoas), mas a deficiência de GH é uma causa menos frequente, afetando apenas cerca entre 3 e 9% das crianças com NF1. Portanto, na maioria das crianças com NF1, a própria doença causa a baixa estatura por mecanismos que ainda não são bem conhecidos.
Como as crianças com NF1 têm risco aumentado para tumores (gliomas e neurofibromas), o uso de tratamento com GH nesta população é controverso e não há consenso ou diretrizes definitivas sobre sua segurança. Embora alguns estudos não mostrem aumento claro do risco de progressão tumoral, a comunidade médica recomenda cautela e avaliação rigorosa caso a caso.
A reposição de hormônio do crescimento (GH) em crianças com NF1 está indicada quando há deficiência de GH documentada por testes de estímulo, manifestando-se como baixa estatura com velocidade de crescimento inadequada.
A deficiência de GH é mais comum naquelas com glioma de vias ópticas (17,8%) do que naquelas sem glioma ( 4,9%). Ver referência [1] abaixo.
Quando investigar a deficiência de GH?
- Crianças com NF1 e baixa estatura (abaixo do percentil 10-25) com velocidade de crescimento inadequada, mesmo na ausência de lesões visíveis na ressonância magnética [2]
- Presença de glioma de vias ópticas, especialmente com envolvimento hipotalâmico ou quiasmático [3][4]
- Sela túrcica vazia parcial ou outras anormalidades hipofisárias [5]
- Sinais de hipopituitarismo (podem incluir puberdade precoce central, síndrome diencefálica ou outras disfunções endócrinas) [3][6]
Diagnóstico
A deficiência de GH deve ser confirmada por testes de estímulo (glucagon, clonidina) com pico de GH menor que 10 ng/mL. É importante notar que a deficiência de GH pode ocorrer mesmo sem lesões suprasselares detectáveis à imagem, sugerindo uma associação intrínseca com a NF1. [5][2]
Segurança da terapia com GH:
A terapia de reposição com GH recombinante em crianças com NF1 e deficiência de GH demonstrou ser eficaz e segura. Dados de 102 crianças com NF1 tratadas com GH mostraram melhora significativa na velocidade de crescimento (de 4,2 cm/ano para 7,1 cm/ano no primeiro ano), sem aumento excessivo do risco de malignidade. A incidência de recorrência ou novos tumores intracranianos foi comparável à esperada em pacientes com NF1 não tratados. [7]
Como é o tratamento?
O tratamento com hormônio de crescimento (GH) é administrado por injeção subcutânea, com formulações diárias ou semanais disponíveis. A dosagem é individualizada e ajustada com base na resposta clínica, níveis de IGF-1 e efeitos colaterais. As injeções são aplicadas por via subcutânea, preferencialmente à noite para ficar semelhante à secreção fisiológica.
A dose é ajustada gradualmente, com consultas a cada 1-2 meses com aumento da dose de 0,1-0,2 mg/dia. Na fase de manutenção as consultas são a cada 6-12 meses.
Cuidados
– Acompanhar os níveis séricos de IGF-1
– Glicemia de jejum e hemoglobina glicada
– Perfil lipídico
– T4 livre (GH aumenta conversão de T4 em T3)
– Função adrenal (GH pode desmascarar insuficiência adrenal central)
– Em crianças: velocidade de crescimento e maturação óssea
Efeitos Colaterais
Em crianças: reações no local da injeção, ginecomastia pré-puberal, dor articular, edema, hipertensão intracraniana benigna, resistência à insulina, progressão de escoliose, epifisiólise femoral proximal
Em adultos: dor articular, cefaleia, síndrome do túnel do carpo, hiperglicemia (mais pronunciados em idosos)
Contraindicações e Precauções
– Câncer ativo é contraindicação absoluta
– Monitorar função tireoidiana e adrenal antes e durante o tratamento
– Ajustar dose de levotiroxina e glicocorticoides conforme necessário
Considerações especiais:
Para crianças com gliomas de vias ópticas estáveis (doença estável, não livre de doença), o início da terapia com GH deve ser discutido com o oncologista [8]
Tradicionalmente, aguarda-se pelo menos 1 ano livre de doença antes de iniciar GH em sobreviventes de tumores malignos, embora craniofaringiomas (tumores benignos) possam ser tratados mais precocemente [8]
A deficiência de GH pode ser transitória em alguns casos, justificando reavaliação [4]
Monitoramento cuidadoso é essencial, incluindo vigilância para epifisiólise femoral proximal e resistência insulínica [8]
O uso de GH em crianças com NF1 sem deficiência de GH não faz sentido pois a causa da baixa estatura não é a deficiência do hormônio e há uma preocupação teórica de que possa facilitar o crescimento tumoral [9]
Acompanhamento:
Recomenda-se seguimento endocrinológico ao longo da vida para todas as crianças com NF1, particularmente aquelas com gliomas de vias ópticas, para identificação precoce de alterações endócrinas secundárias. [6][4][10]
Referências
- Endocrine Manifestations in a Paediatric Cohort of 181 Patients With Neurofibromatosis Type 1. Regala C, Cavaco D, Maciel J, et al. European Journal of Endocrinology. 2025.
- Growth Hormone Deficiency in Children With Neurofibromatosis Type 1 Without Suprasellar Lesions. Vassilopoulou-Sellin R, Klein MJ, Slopis JK. Pediatric Neurology. 2000.
- Pretreatment Endocrine Disorders Due to Optic Pathway Gliomas in Pediatric Neurofibromatosis Type 1: Multicenter Study. Santoro C, Perrotta S, Picariello S, et al. The Journal of Clinical Endocrinology and Metabolism. 2020.
- Endocrine Long-Term Follow-Up of Children With Neurofibromatosis Type 1 and Optic Pathway Glioma Sani I, Albanese A. Hormone Research in Paediatrics. 2017.
- Partial Empty Sella Syndrome, GH Deficiency and Transient Central Adrenal Insufficiency in a Patient With NF1. Kyritsi EM, Hasiotou M, Kanaka-Gantenbein C. Endocrine. 2020.
- Endocrine Implications of Neurofibromatosis 1 in Childhood. Bizzarri C, Bottaro G. Hormone Research in Paediatrics. 2015.
- Growth Hormone Replacement and the Risk of Malignancy in Children With Neurofibromatosis. Howell SJ, Wilton P, Lindberg A, Shalet SM. The Journal of Pediatrics. 1998.
- Hypothalamic-Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. Sklar CA, Antal Z, Chemaitilly W, et al. The Journal of Clinical Endocrinology and Metabolism. 2018.
- Neurofibromatosis 1 in the setting of dual diagnosis: Diagnostic and management conundrums. Muthusamy K, El-Jabali A, Ongie LJ, Dhamija R, Babovic-Vuksanovic D. American Journal of Medical Genetics. Part A. 2022.
- Health Supervision for Children With Neurofibromatosis Type 1. Miller DT, Freedenberg D, Schorry E, et al. Pediatrics. 2019.